Oplysningerne På Webstedet Er Ikke Medicinsk Rådgivning. Vi Sælger Ikke Noget. Nøjagtigheden Af Oversættelsen Er Ikke Garanteret. Ansvarsfraskrivelse
Antipsoriatics, systemiskHumira
Resume af lægemiddeloversigt
Hvad er Humira?
Humira (adalimumab) er et injicerbart protein (antistof), der bruges til behandling af reumatoid arthritis juvenil idiopatisk artritis Psoriasis arthritis ankyloserende spondylitis og plaque psoriasis. Humira bruges også til behandling af Crohns sygdom, efter at andre lægemidler er blevet forsøgt uden vellykket behandling af symptomer.
Hvad er bivirkninger for Humira?
Almindelige bivirkninger af Humira inkluderer
- reaktioner på injektionsstedet (rødme kløe smerter blå hævelse eller blødning)
- hovedpine
- Sæt næse
- sinus smerte eller
- mavesmerter.
Fortæl din læge, hvis du har alvorlige bivirkninger af Humira, herunder:
- Hurtig/uregelmæssig/bankende hjerteslag
- mavesmerter
- blod i afføringen
- Mental/humørændringer
- Alvorlig hovedpine
- let blå mærker eller blødning
- Mørk urin
- gulterende øjne og hud
- bensmerter eller hævelse
- følelsesløshed eller prikken af arme/hænder/ben/fødder
- ustabilitet
- uforklarlig muskelsvaghed
- Sværhedsgrad med at tale/tygge/sluge/ansigtsbevægelser
- Vision ændres
- ekstrem træthed
- ledssmerter eller
- Sommerfuglformet udslæt på næsen og kinderne.
Dosering til Humira
Den anbefalede dosis af HUMIRA til voksne patienter med reumatoid arthritis (RA) psoriasisartrit (PSA) eller ankyloserende spondylitis (AS) administreres 40 mg hver anden uge. Pædiatrisk dosering bestemmes af barnets vægt.
Hvilke stoffer stoffer eller kosttilskud interagerer med Humira?
Andre lægemidler kan interagere med Humira. Fortæl din læge alle receptpligtige og over-the-counter medicin og kosttilskud, du bruger.
Humira During Graviditet og Breastfeeding
Under graviditet bør Humira kun bruges, når det er ordineret. Det er ukendt, om dette stof passerer til modermælk. Lignende medikamenter passerer til modermælk. Kontakt din læge inden amning.
Yderligere oplysninger
Vores Humira (Adalimumab) bivirkninger Drug Center giver et omfattende overblik over tilgængelige lægemiddelinformation om de potentielle bivirkninger, når du tager denne medicin.
FDA -lægemiddelinformation
- Lægemiddelbeskrivelse
- Indikationer
- Dosering
- Bivirkninger
- Lægemiddelinteraktioner
- Advarsler
- Overdosis
- Klinisk farmakologi
- Medicin vejledning
ADVARSEL
Alvorlige infektioner og malignitet
Alvorlige infektioner
Patienter, der behandles med Humira, har en øget risiko for at udvikle alvorlige infektioner, der kan føre til indlæggelse eller død [se advarsler og FORHOLDSREGLER ]. De fleste patienter, der udviklede disse infektioner, tog samtidig immunsuppressiva, såsom methotrexat eller kortikosteroider.
Afbryd Humira, hvis en patient udvikler en alvorlig infektion eller sepsis.
Rapporterede infektioner inkluderer:
- Aktiv tuberkulose (TB) inklusive reaktivering af latent TB. Patienter med TB har ofte præsenteret for formidlet eller ekstrapulmonal sygdom. Test patienter for latent TB før Humira -brug og under terapi. Initier behandling af latent TB inden Humira -brug.
- Invasive svampeinfektioner inklusive histoplasmosis coccidioidomycosis candidiasis aspergillosis blastomycosis og pneumocystose. Patienter med histoplasmose eller andre invasive svampeinfektioner kan have formidlet snarere end lokal sygdom. Antigen- og antistofprøvning til histoplasmose kan være negativ hos nogle patienter med aktiv infektion. Overvej empirisk antisvampetapi hos patienter, der er i fare for invasive svampeinfektioner, der udvikler alvorlig systemisk sygdom.
- Bakteriel viral og andre infektioner på grund af opportunistiske patogener inklusive Legionella og Listeria.
Overvej omhyggeligt risikoen og fordelene ved behandling med HUMIRA, inden behandling af terapi hos patienter med kronisk eller tilbagevendende infektion.
Overvåg patienter nøje for udviklingen af tegn og symptomer på infektion under og efter behandling med HUMIRA inklusive den mulige udvikling af TB hos patienter, der testede negativt for latent TB -infektion, inden de indledte terapi [se advarsler og FORHOLDSREGLER og Bivirkninger ].
Malignitet
Lymfom og andre maligniteter Nogle dødelige er rapporteret hos børn og unge patienter behandlet med TNF -blokkeere, herunder Humira [se advarsler og FORHOLDSREGLER ]. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL) a rare type of T-cell lymphoma have been reported in patients treated with TNF blockers including Humira. These cases have had a very aggressive disease course og have been fatal. The majority of reported TNF blocker cases have occurrød in patients with Crohn's disease or ulcerative colitis og the majority were in adolescent og young adult males. Almost all these patients had received treatment with azathioprine or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants [see ADVARSELS AND FORHOLDSREGLER ].
Beskrivelse for Humira
Humira (adalimumab) er en rekombinant human IgG1 monoklonalt antistof specifikt for human tumor nekrose faktor (TNF). Humira blev oprettet ved hjælp af fagdisplay -teknologi, der resulterede i et antistof med human afledt tunge og lette kæde -variable regioner og human IgG1: K konstante regioner. Adalimumab produceres ved rekombinant DNA -teknologi i et pattedyrscelleekspressionssystem og renses ved en proces, der inkluderer specifik viral inaktiverings- og fjernelsestrin. Det består af 1330 aminosyrer og har en molekylvægt på ca. 148 kilodaltons.
Humira leveres som en steril konserveringsfri opløsning af adalimumab til subkutan administration. Lægemiddelproduktet leveres som enten en engangs-præfyldt pen (Humira Pen) som en engangsudfyldt glassprøjtning på 1 ml eller som en enkeltbrugsinstitutionel brugshætteglas. Lukket inden i pennen er en engangsudfyldt glassprøjte 1 ml. Løsningen af Humira er klar og farveløs med en pH på ca. 5,2.
Hver 80 mg/0,8 ml -præfyldt sprøjte eller -forparet pen leverer 0,8 ml (80 mg) lægemiddelprodukt. Hver 0,8 ml Humira indeholder adalimumab (80 mg) mannitol (NULL,6 mg) polysorbat 80 (NULL,8 mg) og vand til injektion USP.
Hver 40 mg/0,4 ml -præfyldt sprøjte eller -forfyldt pen leverer 0,4 ml (40 mg) lægemiddelprodukt. Hver 0,4 ml Humira indeholder adalimumab (40 mg) mannitol (NULL,8 mg) polysorbat 80 (NULL,4 mg) og vand til injektion USP.
Hver 40 mg/0,8 ml forfyldt sprøjteforfyldt pen eller institutionel brug af engangsbrug leverer 0,8 ml (40 mg) lægemiddelprodukt. Hver 0,8 ml Humira indeholder adalimumab (40 mg) citronsyre monohydrat (NULL,04 mg) dibasisk natriumphosphatdihydrat (NULL,22 mg) mannitol (NULL,6 mg) monobasisk sodium phosphatdihydrat (NULL,69 mg) polysorbate 80 (NULL,8 mg) Sodium phlorid (NULL,9 Mg) natriumcitrat (NULL,24 mg) og vand til injektion USP. Natriumhydroxid tilsættes som nødvendigt for at justere pH.
Hver 20 mg/0,2 ml præfyldt sprøjte leverer 0,2 ml (20 mg) lægemiddelprodukt. Hver 0,2 ml Humira indeholder adalimumab (20 mg) mannitol (NULL,4 mg) polysorbat 80 (NULL,2 mg) og vand til injektion USP.
Hver 20 mg/0,4 ml præfyldt sprøjte leverer 0,4 ml (20 mg) lægemiddelprodukt. Hver 0,4 ml Humira indeholder adalimumab (20 mg) citronsyre -monohydrat (NULL,52 mg) dibasisk natriumphosphatdihydrat (NULL,61 mg) mannitol (NULL,8 mg) monobasisk chlorid -dihydrat (NULL,34 mg) polysorbate 80 (NULL,4 mg) Sodium phosphatdihydrat (NULL,34 mg) Polysorbate 80 (NULL,4 mg) Sodium -chlorid (2.47777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777777 havde Polysorbate Mg) natriumcitrat (NULL,12 mg) og vand til injektion USP. Natriumhydroxid tilsættes som nødvendigt for at justere pH.
Hver 10 mg/0,1 ml præfyldt sprøjte leverer 0,1 ml (10 mg) lægemiddelprodukt. Hver 0,1 ml Humira indeholder adalimumab (10 mg) mannitol (NULL,2 mg) polysorbat 80 (NULL,1 mg) og vand til injektion USP.
Hver 10 mg/0,2 ml præfyldt sprøjte leverer 0,2 ml (10 mg) lægemiddelprodukt. Hver 0,2 ml Humira indeholder adalimumab (10 mg) citronsyre monohydrat (NULL,26 mg) dibasisk natriumphosphatdihydrat (NULL,31 mg) mannitol (NULL,4 mg) monobasisk chlorid -dihydrat (NULL,17 mg) polysorbate 80 (NULL,2 mg) Sodium phlorid (NULL,2 Mg) natriumcitrat (NULL,06 mg) og vand til injektion USP. Natriumhydroxid tilsættes som nødvendigt for at justere pH.
Bruger til Humira
Reumatoid arthritis
Humira er indikeret til reduktion af tegn og symptomer, der inducerer større klinisk respons, der hæmmer udviklingen af strukturel skade og forbedrer fysisk funktion hos voksne patienter med moderat til alvorligt aktiv reumatoid arthritis. Humira kan bruges alene eller i kombination med methotrexat eller anden ikke-biologisk sygdomsmodificerende anti-rheumatiske lægemidler (DMARD'er).
Juvenil idiopatisk arthritis
Humira er indikeret til reduktion af tegn og symptomer på moderat til alvorligt aktiv polyartikulær juvenil idiopatisk arthritis hos patienter 2 år og ældre. Humira kan bruges alene eller i kombination med methotrexat.
Psoriasis arthritis
Humira er indikeret til reduktion af tegn og symptomer, der hæmmer udviklingen af strukturel skade og forbedrer fysisk funktion hos voksne patienter med aktiv psoriasisartrit. Humira kan bruges alene eller i kombination med ikke-biologiske dmards.
Ankyloserende spondylitis
Humira er indikeret til reduktion af tegn og symptomer hos voksne patienter med aktiv ankyloserende spondylitis.
Crohns sygdom
Humira er indikeret til behandling af moderat til alvorligt aktiv Crohns sygdom hos voksne og pædiatriske patienter 6 år og ældre.
Ulcerøs colitis
Humira er indikeret til behandling af moderat til alvorligt aktiv ulcerøs colitis hos voksne og pædiatriske patienter 5 år og ældre.
Begrænsninger af brug
Effektiviteten af Humira er ikke blevet fastlagt hos patienter, der har mistet responsen på eller var intolerante over for TNF -blokkeere [se Kliniske studier ].
Plaque psoriasis
Humira er indikeret til behandling af voksne patienter med moderat til svær kronisk plaque -psoriasis, der er kandidater til systemisk terapi eller fototerapi, og når andre systemiske terapier er medicinsk mindre passende. Humira bør kun administreres til patienter, der vil blive nøje overvåget og have regelmæssige opfølgningsbesøg med en læge [se ADVARSELS AND FORHOLDSREGLER ].
Hidradenitis suppurativ
Humira er indikeret til behandling af moderat til svær hidradenitis suppurativa hos patienter 12 år og ældre.
Uveitis
Humira er indikeret til behandling af ikke-infektiøst mellemliggende posterior og panuveitis hos voksne og pædiatriske patienter 2 år og ældre.
Dosering til Humira
Reumatoid arthritis Psoriasis arthritis And Ankyloserende spondylitis
Den anbefalede subkutane dosering af HUMIRA til voksne patienter med reumatoid arthritis (RA) psoriasisartrit (PSA) eller ankyloserende spondylitis (AS) administreres 40 mg hver anden uge. Methotrexat (MTX) Andre ikke-biologiske DMARDS-glukokortikoider ikke-steroide antiinflammatoriske lægemidler (NSAID'er) og/eller smertestillende midler kan fortsættes under behandling med Humira. I behandlingen af RA kan nogle patienter, der ikke tager samtidig MTX, få yderligere fordele ved at øge doseringen af Humira til 40 mg hver uge eller 80 mg hver anden uge.
Juvenil idiopatisk arthritis Or Pædiatrisk uveitis
Den anbefalede subkutane dosering af HUMIRA for patienter 2 år og ældre med polyartikulær juvenil idiopatisk arthritis (JIA) eller pædiatrisk uveitis er baseret på vægt som vist nedenfor. MTX -glukokortikoider NSAID'er og/eller smertestillende midler kan fortsættes under behandling med Humira.
| Pædiatrisk vægt (2 år gammel og ældre) | Anbefalet dosering |
| 10 kg (22 lbs) til mindre end 15 kg (33 lbs) | 10 mg hver anden uge |
| 15 kg (33 lbs) til mindre end 30 kg (66 lbs) | 20 mg hver anden uge |
| 30 kg (66 lbs) og større | 40 mg hver anden uge |
Humira er ikke undersøgt hos patienter med polyartikulær JIA eller pædiatrisk uveitis mindre end 2 år eller hos patienter med en vægt under 10 kg.
Crohns sygdom
Voksne
Den anbefalede subkutan dosering af Humira til voksne patienter med Crohns sygdom (CD) er 160 mg oprindeligt på dag 1 (givet på en dag eller splittet over to på hinanden følgende dage) efterfulgt af 80 mg to uger senere (dag 15). To uger senere (dag 29) begynder en dosering på 40 mg hver anden uge. Aminosalicylater og/eller kortikosteroider kan fortsættes under behandling med Humira. Azathioprin 6-mercaptopurine (6-mp) [se ADVARSELS AND FORHOLDSREGLER ] eller MTX kan fortsættes under behandling med Humira om nødvendigt.
Pædiatri
Den anbefalede subkutane dosering af Humira til pædiatriske patienter 6 år og ældre med Crohns sygdom (CD) er baseret på kropsvægt som vist nedenfor:
| Pædiatrisk vægt | Anbefalet dosering | |
| Dage 1 til 15 | Fra dag 29 | |
| 17 kg (37 lbs) til mindre end 40 kg (88 lbs) | Dag 1: 80 mg dag 15: 40 mg | 20 mg hver anden uge |
| 40 kg (88 lbs) og større | Dag 1: 160 mg (enkelt dosis eller splittet over to på hinanden følgende dage) Dag 15: 80 mg | 40 mg hver anden uge |
Ulcerøs colitis
Voksne
Den anbefalede subkutan dosering af Humira til voksne patienter med ulcerøs colitis er 160 mg oprindeligt på dag 1 (givet på en dag eller splittet over to på hinanden følgende dage) efterfulgt af 80 mg to uger senere (dag 15). To uger senere (dag 29) fortsætter med en dosering på 40 mg hver anden uge.
Afbryd Humira hos voksne patienter uden bevis for klinisk remission med otte uger (dag 57) af terapi. Aminosalicylater og/eller kortikosteroider kan fortsættes under behandling med Humira. Azathioprin og 6-mercaptopurine (6-mp) [se ADVARSELS AND FORHOLDSREGLER ] kan fortsættes under behandling med Humira om nødvendigt.
Pædiatri
Den anbefalede subkutan dosering af HUMIRA til pædiatriske patienter 5 år og ældre med ulcerøs colitis er baseret på kropsvægt som vist nedenfor:
| Pædiatrisk vægt | Anbefalet dosering | |
| Dage 1 til 15 | Fra dag 29* | |
| 20 kg (44 lbs) til mindre end 40 kg (88 lbs) | Dag 1: 80 mg Dag 8: 40 mg Dag 15: 40 mg | 40 mg hver anden uge or 20 mg every week |
| 40 kg (88 lbs) og større | Dag 1: 160 mg (enkelt dosis eller split over to på hinanden følgende dage) Dag 8: 80 mg Dag 15: 80 mg | 80 mg hver anden uge eller 40 mg hver uge |
| * Fortsæt den anbefalede pædiatriske dosering hos patienter, der bliver 18 år gammel, og som er godt kontrolleret på deres Humira-regime. |
Plaque psoriasis Or Voksen uveitis
Den anbefalede subkutane dosering af HUMIRA til voksne patienter med plaque psoriasis (PS) eller uveitis (UV) er en indledende dosis på 80 mg efterfulgt af 40 mg givet hver anden uge startende en uge efter den indledende dosis. Brugen af Humira i moderat til svær kronisk PS ud over et år er ikke blevet evalueret i kontrollerede kliniske studier.
Hidradenitis suppurativ
Voksne
Den anbefalede subkutan dosering af HUMIRA til voksne patienter med hidradenitis suppurativa (HS) er en indledende dosis på 160 mg (givet på en dag eller splittet over to på hinanden følgende dage) efterfulgt af 80 mg to uger senere (dag 15). Begynd 40 mg ugentligt eller 80 mg hver anden uge dosering to uger senere (dag 29).
Unge
Den anbefalede subkutan dosering af Humira til unge patienter 12 år og ældre, der vejer mindst 30 kg med hidradenitis suppurativa (HS) er baseret på kropsvægt som vist nedenfor [se Brug i specifikke populationer og Klinisk farmakologi ]:
| Kropsvægt hos unge patienter (12 år gammel og ældre) | Anbefalet dosering |
| 30 kg (66 lbs) til mindre end 60 kg (132 lbs) |
|
| 60 kg (132 lbs) og større |
|
Overvågning for at vurdere sikkerheden
Før man påbegyndte Humira og med jævne mellemrum under terapi evaluerer patienter for aktiv tuberkulose og test for latent infektion [se ADVARSELS AND FORHOLDSREGLER ].
Generelle overvejelser for administration
Humira er beregnet til brug under vejledning og overvågning af en læge. En patient kan selv indlede Humira eller en plejeperson kan injicere Humira ved hjælp af enten Humira-pen eller forudfyldt sprøjte, hvis en læge bestemmer, at den er passende og med medicinsk opfølgning efter behov efter korrekt træning i subkutan injektionsteknik.
Humira kan tages ud af køleskabet i 15 til 30 minutter, før den injicerer for at lade væsken komme til stuetemperatur. Fjern ikke hætten eller dækslet, mens den tillader det at nå stuetemperatur. Undersøg omhyggeligt opløsningen i Humira Pen-præfyldt sprøjte eller enkeltdosis institutionel brug hætteglas til partikler og misfarvning inden subkutan administration. Hvis der ikke er anført partikler og misfarvninger, skal du ikke bruge produktet. Humira indeholder ikke konserveringsmidler; Kasser derfor ubrugte dele af lægemiddel tilbage fra sprøjten. Bemærk: Instruer patienter, der er følsomme over for latex, om ikke at håndtere nåledækslet på Humira 40 mg/0,8 ml pen og 40 mg/0,8 ml 20 mg/0,4 ml og 10 mg/0,2 ml præfyldt sprøjte, fordi den kan indeholde naturlig gummi latex [se Hvor leveret / Opbevaring og håndtering ].
Instruer patienter, der bruger Humira -pen eller forudfyldt sprøjte til at injicere det fulde beløb i sprøjten i henhold til anvisningerne i instruktionerne til brug [se Brug til brug ].
Injektioner skal forekomme på separate steder i låret eller maven. Drej injektionssteder og giver ikke injektioner i områder, hvor huden er mørt mærket rød eller hårdt .â
Humira-enkeltdosis institutionel brug hætteglas er kun til administration inden for en institutionel ramme, såsom en hospitalets læges kontor eller klinik. Træk dosis tilbage ved hjælp af en steril nål og sprøjte og administrerer hurtigt af en sundhedsudbyder inden for en institutionel indstilling. Administrer kun en dosis pr. Hætteglas. Hætteglasset indeholder ikke konserveringsmidler; Kasser derfor ubrugte dele.
Hvor leveret
Dosering Forms And Strengths
Humira er en klar og farveløs løsning tilgængelig som:
- Pen (Humira Pen)
- Injektion: 80 mg/0,8 ml i en enkeltdosis pen.
- Injektion: 40 mg/0,8 ml i en enkeltdosis pen.
- Injektion: 40 mg/0,4 ml i en enkeltdosis pen.
- Foretfyldt sprøjte
- Injektion: 80 mg/0,8 ml i en enkelt dosis forfyldt glassprøjte.
- Injektion: 40 mg/0,8 ml i en enkeltdosis forfyldt glassprøjte.
- Injektion: 40 mg/0,4 ml i en enkeltdosis forfyldt glassprøjte.
- Injektion: 20 mg/0,4 ml i en enkeltdosis forfyldt glassprøjte.
- Injektion: 20 mg/0,2 ml i en enkeltdosis forfyldt glassprøjte.
- Injektion: 10 mg/0,2 ml i en enkeltdosis forfyldt glassprøjte.
- Injektion: 10 mg/0,1 ml i en enkeltdosis forfyldt glassprøjte.
- Enkeltdosis institutionel brug hætteglas
- Injektion: 40 mg/0,8 ml i et enkelt dosis glashætteglas til institutionel brug.
Opbevaring og håndtering
Humira® (adalimumab) leveres som en konserveringsfri steril klar og farveløs løsning til subkutan administration. Følgende emballagekonfigurationer er tilgængelige.
- Humira Pen Carton - 40 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-4339-02.
- Humira Pen Carton - 40 mg/0,4 ml
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0554-02.
- Humira Pen Carton †80 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0124-02.
- Humira Pen 40 mg/0.8 mL - Starter Package for Crohn's Disease Ulcerøs colitis or Hidradenitis suppurativ
- Humira is supplied in a carton containing 6 alcohol preps og 6 dose trays (Starter Package for Crohns sygdom Ulcerøs colitis or Hidradenitis suppurativ). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-4339-06.
- Humira Pen 40 mg/0,4 ml - Starter Package for Crohn's Disease Ulcerøs colitis or Hidradenitis suppurativ
- Humira is supplied in a carton containing 6 alcohol preps og 6 dose trays (Starter Package for Crohns sygdom Ulcerøs colitis or Hidradenitis suppurativ). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0554-06.
- Humira Pen 80 mg/0.8 mL - Starter Package for Crohn's Disease Ulcerøs colitis or Hidradenitis suppurativ
- Humira is supplied in a carton containing 4 alcohol preps og 3 dose trays (Starter Package for Crohns sygdom Ulcerøs colitis or Hidradenitis suppurativ). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0124-03.
- Humira Pen 40 mg/0.8 mL - Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package
- Humira is supplied in a carton containing 4 alcohol preps og 4 dose trays (Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-4339-07.
- Humira Pen 40 mg/0,4 ml - Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package
- Humira is supplied in a carton containing 4 alcohol preps og 4 dose trays (Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0554-04.
- Humira Pen 80 mg/0.8 mL og 40 mg/0,4 ml - Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package
- Humira is supplied in a carton containing 4 alcohol preps og 3 dose trays (Psoriasis Uveitis or Adolescent Hidradenitis suppurativ Starter Package). One dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The other two dose trays each consist of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Antal er 0074-1539-03.
- Humira Pen 80 mg/0.8 mL †Starter Package for Pædiatrisk ulcerøs colitis (4 count)
- Humira is supplied in a carton containing 4 alcohol preps og 4 dose trays (Starter Package for Pædiatrisk ulcerøs colitis). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0124-04.
- Foretfyldt sprøjte Carton - 40 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-3799-02.
- Foretfyldt sprøjte Carton - 40 mg/0,4 ml
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0243-02.
- Foretfyldt sprøjte Carton - 20 mg/0.4 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 20 mg/0.4 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-9374-02.
- Foretfyldt sprøjte Carton - 20 mg/0.2 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 20 mg/0.2 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0616-02.
- Foretfyldt sprøjte Carton - 10 mg/0.2 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 10 mg/0.2 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-6347-02.
- Foretfyldt sprøjte Carton - 10 mg/0.1 mL
- Humira is supplied in a carton containing two alcohol preps og two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 10 mg/0.1 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-0817-02.
- Humira Foretfyldt sprøjte 40 mg/0.8 mL - Pædiatrisk Crohns sygdom Starter Package (6 count)
- Humira is supplied in a carton containing 6 alcohol preps og 6 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-3799-06.
- Humira Foretfyldt sprøjte 80 mg/0.8 mL - Pædiatrisk Crohns sygdom Starter Package (3 count)
- Humira is supplied in a carton containing 4 alcohol preps og 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Nummer er 0074-2540-03.
- Humira Foretfyldt sprøjte 40 mg/0.8 mL - Pædiatrisk Crohns sygdom Starter Package (3 count)
- Humira is supplied in a carton containing 4 alcohol preps og 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural gnideber latex. The NDC Nummer er 0074-3799-03.
- Humira Foretfyldt sprøjte 80 mg/0.8 mL og 40 mg/0,4 ml - Pædiatrisk Crohns sygdom Starter Package (2 count)
- Humira is supplied in a carton containing 2 alcohol preps og 2 dose trays (Pediatric Starter Package). One dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The other dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ikke made with natural gnideber latex. The NDC Antal er 0074-0067-02.
- Enkeltdosis institutionel brug hætteglas Carton - 40 mg/0.8 mL
- Humira is supplied for institutional use only in a carton containing a single-dose glass vial providing 40 mg/0.8 mL of Humira. The vial stopper is ikke made with natural gnideber latex. The NDC Nummer er 0074-3797-01.
Opbevaring og stabilitet
Brug ikke ud over udløbsdatoen på containeren. Humira skal køles ved 36 ° F til 46 ° F (2 ° C til 8 ° C). Frys ikke. Brug ikke, hvis det er frosset, selvom det er optøet.
Opbevares i original karton indtil administrationstidspunktet for at beskytte mod lys.
Om nødvendigt for eksempel når Humira rejser kan opbevares ved stuetemperatur op til maksimalt 25 ° C (25 ° C) i en periode på op til 14 dage med beskyttelse mod lys. Humira bør kasseres, hvis det ikke bruges inden for den 14-dages periode. Registrer datoen, hvor Humira først fjernes fra køleskabet i de rum, der er leveret på karton- og dosisbakken.
Opbevar ikke Humira i ekstrem varme eller kulde.
Abbvie Inc. North Chicago IL 60064 U.S.A. US License Number. Revideret: Feb 2021
Bivirkninger for Humira
Følgende klinisk signifikante bivirkninger er beskrevet andetsteds i mærkningen:
- Alvorlige infektioner [see ADVARSELS AND FORHOLDSREGLER ]
- Maligniteter [se ADVARSELS AND FORHOLDSREGLER ]
- Overfølsomhedsreaktioner [se ADVARSELS AND FORHOLDSREGLER ]
- Hepatitis B -virusreaktivering [se ADVARSELS AND FORHOLDSREGLER ]
- Neurologiske reaktioner [se ADVARSELS AND FORHOLDSREGLER ]
- Hæmatologiske reaktioner [se ADVARSELS AND FORHOLDSREGLER ]
- Hjertesvigt [se ADVARSELS AND FORHOLDSREGLER ]
- Autoimmunity [se ADVARSELS AND FORHOLDSREGLER ]
Kliniske forsøg oplever
Fordi kliniske forsøg udføres under vidt forskellige tilstande, kan der ikke sammenlignes bivirkninger, der er observeret i de kliniske forsøg med et lægemiddel, ikke direkte med hastigheder i de kliniske forsøg med et andet lægemiddel og muligvis ikke afspejler de satser, der er observeret i praksis.
Den mest almindelige bivirkning med Humira var injektionsstedets reaktioner. I placebokontrollerede forsøg udviklede 20% af patienterne, der blev behandlet med Humira, injektionsstedets reaktioner (erythema og/eller kløe blødningssmerter eller hævelse) sammenlignet med 14% af patienterne, der fik placebo. De fleste reaktioner på injektionsstedet blev beskrevet som milde og krævede generelt ikke ophør med lægemiddel.
Andelen af patienter, der afbrød behandlingen på grund af bivirkninger under den dobbeltblinde placebokontrollerede del af undersøgelser hos patienter med RA (dvs. undersøgelser RA-I RAII RA-III og RA-IV) var 7% for patienter, der tog Humira og 4% for placebo-behandlede patienter. De mest almindelige bivirkninger, der førte til seponering af HUMIRA i disse RA -undersøgelser, var klinisk flare -reaktion (NULL,7%) udslæt (NULL,3%) og lungebetændelse (NULL,3%).
Infektioner
I de kontrollerede dele af de 39 globale Humira-kliniske forsøg hos voksne patienter med RA PSA som CD UC PS HS og UV var hastigheden af alvorlige infektioner 4,3 pr. 100 patientår hos 7973 HUMIRA-behandlede patienter i forhold til en hastighed på 2,9 pr. 100 patientår i 4848 kontrolbehandlede patienter. Alvorlige infektioner observeret inkluderede lungebetændelse septisk arthritis protese og postkirurgiske infektioner erysipelas cellulitis diverticulitis og pyelonephritis [se ADVARSELS AND FORHOLDSREGLER ].
Tuberkulose og opportunistiske infektioner
I 52 globale kontrollerede og ukontrollerede kliniske forsøg i RA PSA som CD UC PS HS og UV, der inkluderede 24605 HUMIRA-behandlede patienter, var hastigheden for rapporteret aktiv tuberkulose 0,20 pr. 100 patientår og hastigheden af positiv PPD-konvertering var 0,09 pr. 100 patientår. I en undergruppe på 10113 U.S. og canadiske HUMIRA-behandlede patienter var hastigheden for rapporteret aktiv TB 0,05 pr. 100 patientår og hastigheden for positiv PPD-konvertering var 0,07 pr. 100 patientår. Disse forsøg omfattede rapporter om miliær lymfatisk peritoneal og lunge TB. ADVARSELS AND FORHOLDSREGLER ].
Autoantistoffer
I reumatoid arthritis kontrollerede forsøg 12% af patienterne behandlet med Humira og 7% af placebo -behandlede patienter, der havde negative baseline -ANA -titere, udviklede positive titere i uge 24. To patienter ud af 3046 behandlet med HUMIRA udviklede kliniske tegn, der sørger for newonset lupus -ligner syndrom. Patienterne forbedrede sig efter seponering af terapi. Ingen patienter udviklede lupusnefritis eller symptomer på centralnervesystemet. Virkningen af langvarig behandling med Humira på udviklingen af autoimmune sygdomme er ukendt.
Leverenzymhøjder
Der har været rapporter om alvorlige leverreaktioner, herunder akut leversvigt hos patienter, der modtager TNF-blokkere. I kontrollerede fase 3-forsøg med HUMIRA (40 mg SC hver anden uge) hos patienter med RA PSA og som med kontrolperioden varighed fra 4 til 104 ugers ELT-forhøjninger ≥ 3 x ULN forekom hos 3,5% af HUMIRA-behandlede patienter og 1,5% af kontrollerede patienter. Da mange af disse patienter i disse forsøg også tog medicin, der forårsager leverenzymhøjder (f.eks. NSAIDS MTX), er forholdet mellem Humira og leverenzymhøjderne ikke klare. I et kontrolleret fase 3-forsøg med HUMIRA hos patienter med polyartikulær JIA, der var 4 til 17 år ALT-forhøjninger ≥ 3 x ULN, forekom hos 4,4% af HUMIRA-behandlede patienter og 1,5% af kontrolbehandlede patienter (ALT mere almindeligt end AST); Leverenzymtestforhøjelser var hyppigere blandt dem, der blev behandlet med kombinationen af HUMIRA og MTX end dem, der blev behandlet med Humira alene. Generelt førte disse højder ikke til ophør af Humira -behandling. Ingen ALT-forhøjninger ≥ 3 x Uln forekom i open-label-undersøgelsen af Humira hos patienter med polyartikulær JIA, der var 2 til <4 years.
I kontrollerede fase 3-forsøg med HUMIRA (indledende doser på 160 mg og 80 mg eller 80 mg og 40 mg på henholdsvis dag 1 og 15 efterfulgt af 40 mg hver anden uge) hos voksne patienter med Crohns sygdom med en kontrolperiode varierede fra 4 til 52 ugers ELT-forhøjelser ≥ 3 X Uln forekom hos 0,9% af Humira-behandlingen og 0,9% af kontrol-trimatpatienter. I fase 3 -forsøget med HUMIRA hos pædiatriske patienter med Crohns sygdom, der evaluerede effektiviteten og sikkerheden af to kropsvægtbaserede vedligeholdelsesdosisregimer efter kropsvægtbaseret induktionsterapi op til 52 ugers behandlings -ALT -forhøjninger ≥ 3 x ULN forekom i 2,6% (5/192) af patienter af hvem 4 modtog contriske immunosuppessanter ved basis; Ingen af disse patienter ophørte på grund af abnormiteter i ALT -tests. I kontrollerede fase 3-forsøg med HUMIRA (indledende doser på 160 mg og 80 mg på dag 1 og 15 efterfulgt af 40 mg hver anden uge) hos voksne patienter med UC med kontrolperiode varighed fra 1 til 52 ugers ALT-forhøjninger ≥3 x Uln forekom hos 1,5% af HUMIRA-behandlingspatienter og 1,0% af kontrolbehandlede patienter. I det kontrollerede fase 3 -forsøg med HUMIRA hos patienter med pædiatrisk ulcerøs colitis (n = 93), som evaluerede effektivitet og sikkerhed for en vedligeholdelsesdosis på 0,6 mg/kg (maksimal 40 mg) hver anden uge (n = 31) og en vedligeholdelsesdosis på 0,6 mg/kg (maksimal 40 mg) hver uge (n = 32) efter kropsvægtbaseret induktion doser på 2,4 mg/kg (maksimum på 40 mg) hver uge (n = 32) efter kropsvægtbaseret induktion doser doses på 2,4 mg/kg (maksimum på 40 mg) (n = n = 32 Mg) i uge 0 og uge 1 og 1,2 mg/kg (maksimalt 80 mg) ved uge 2 (n = 63) eller en induktionsdosis på 2,4 mg/kg (maksimum på 160 mg) ved uge 0 placebo i uge 1 og 1,2 mg/kg (maks. 80 mg) ved uge 2 (n = 30) ALT -ELEETATIONS ≥ 3 X ULN Uln forekom i 1,1% (1/93). I kontrollerede fase 3-forsøg med HUMIRA (initial dosis på 80 mg derefter 40 mg hver anden uge) hos patienter med PS med kontrolperiode varighed fra 12 til 24 ugers ELT-forhøjninger ≥ 3 x ULN forekom hos 1,8% af HUMIRA-behandlede patienter og 1,8% af kontrolbehandlede patienter. I kontrollerede forsøg med HUMIRA (indledende doser på 160 mg i uge 0 og 80 mg i uge 2 efterfulgt af 40 mg hver uge fra uge 4) i personer med HS med en kontrolperiode varighed fra 12 til 16 ugers ALT-forhøjninger ≥ 3 x Uln forekom i 0,3% af Humira-behandlingspersoner og 0,6% af kontrolbehandlingsemner. I kontrollerede forsøg med HUMIRA (indledende doser på 80 mg i uge 0 efterfulgt af 40 mg hver anden uge, der starter ved uge 1) hos voksne patienter med uveitis med en eksponering på 165,4 Pys og 119,8 Pys i HUMIRA-behandlede og kontrol-behandlede patienter med henholdsvis ELT-forhøjelser ≥ 3 X Uln i 2,4% af HUMIRA-behandlede og 2,4% kontrol-T-TEAT-ENT-ELTATIONS ≥ 3 X ULN ULN i 2,4% af HUMIRA-behandlede og 2,4% kontrol-T-TEATEDET-ENT-ENT-ELTATIONS ≥ 3 X Uln.
Andre bivirkninger
Reumatoid arthritis Kliniske studier
Dataene beskrevet nedenfor afspejler eksponering for Humira hos 2468 patienter, herunder 2073 udsat i 6 måneder 1497 udsat for mere end et år og 1380 i tilstrækkelige og godt kontrollerede studier (undersøgelser RA-I RA-II RA-III og RA-IV). Humira blev primært undersøgt i placebokontrollerede forsøg og i langvarige opfølgningsundersøgelser i op til 36 måneders varighed. Befolkningen havde en gennemsnitlig alder på 54 år 77% var kvindelige 91% var kaukasiske og havde moderat til alvorligt aktiv reumatoid arthritis. De fleste patienter modtog 40 mg Humira hver anden uge [se Kliniske studier ].
Tabel 1 opsummerer reaktioner rapporteret med en hastighed på mindst 5% hos patienter behandlet med Humira 40 mg hver anden uge sammenlignet med placebo og med en forekomst højere end placebo. I undersøgelsen RA-III var typerne og frekvenserne af bivirkninger i det andet år open-label-udvidelse svarende til dem, der blev observeret i det etårige dobbeltblinde del.
Tabel 1: Bivirkninger rapporteret af ≥5% af patienterne behandlet med Humira i placebo-kontrolleret periode med samlede RA-undersøgelser (undersøgelser RA-I RA-II RA-III og RA-IV)
| Humira 40 mg subcutaneous Every Andre Week (N = 705) | Placebo (N = 690) | |
| Bivirkning (foretrukket udtryk) | ||
| Åndedrætsværn | ||
| Øvre luftvejsinfektion | 17% | 13% |
| Bihulebetændelse | 11% | 9% |
| Influenza syndrom | 7% | 6% |
| Gastrointestinal | ||
| Kvalme | 9% | 8% |
| Mavesmerter | 7% | 4% |
| Laboratorietest* | ||
| Laboratorietest unormal | 8% | 7% |
| Hypercholesterolæmi | 6% | 4% |
| Hyperlipidæmi | 7% | 5% |
| Hæmaturi | 5% | 4% |
| Alkalisk phosphatase steg | 5% | 3% |
| Andre | ||
| Hovedpine | 12% | 8% |
| Udslæt | 12% | 6% |
| Utilsigtet skade | 10% | 8% |
| Reaktion på injektionsstedet ** | 8% | 1% |
| Rygsmerter | 6% | 4% |
| Urinvejsinfektion | 8% | 5% |
| Hypertension | 5% | 3% |
| * Laboratorietest abnormiteter blev rapporteret som bivirkninger i europæiske forsøg ** Inkluderer ikke injektionsstedet erythema kløe blødningssmerter eller hævelse |
Mindre almindelige bivirkninger i reumatoid arthritis kliniske undersøgelser
Andre infrequent serious adverse reactions that Gør ikke appear in the Warnings og Precautions or Adverse Reaction sections that occurrød at an incidence of less than 5% in Humira-treated patients in RA studies were:
Krop som helhed: Smerter i ekstremitet
Kardiovaskulært system: Arytmi Atrieflimmer Brystsmerter Koronar arterieforstyrrelse Hjertestop hypertensiv encephalopati Myokardieinfarkt Pericardial effusion Pericarditis Synkope Tachycardia
Fordøjelsessystem: Cholecystitis cholelithiasis esophagitis gastroenteritis gastrointestinal blødning hepatisk nekrose opkast
Endokrin system: Parathyroidforstyrrelse
Hemisk og lymfatisk system: Agranulocytose polycythemia
Metaboliske og ernæringsmæssige lidelser: Dehydrering Healing Abnormal ketose Paraproteinæmi Perifert ødem
Muskulo-skelet-system: Arthritis knoglerforstyrrelse knoglemraktur (ikke spontan) knoglernekrose ledforstyrrelse muskelkramper myasthenia pyogen arthritis synovitis seneforstyrrelse
Neoplasia: Adenom
Nervesystem: Forvirring Paræstesi Subdural Hematoma Tremor
Åndedrætsværn System: Astma bronchospasme dyspnø -lungefunktion nedsat pleural effusion
Særlige sanser: Grå stær
Trombose: Thrombosis ben
Urogenitalt system: Cystitis nyreberegning menstruationsforstyrrelse
Juvenil idiopatisk arthritis Kliniske studier
Generel Kliniske studier ] var ens i frekvens og type som dem, der blev set hos voksne patienter [se ADVARSELS AND FORHOLDSREGLER Bivirkninger ]. Important findings og differences from adults are discussed in the following paragraphs.
I undersøgelsen blev Jia-I Humira undersøgt hos 171 patienter, der var 4 til 17 år med polyartikulær JIA. Alvorlige bivirkninger rapporteret i undersøgelsen omfattede neutropeni -streptokokk pharyngitis øgede aminotransferaser herpes zoster myositis metrorragia og blindtarmbetændelse. Alvorlige infektioner blev observeret hos 4% af patienterne inden for ca. 2 års påbegyndelse af behandling med Humira og omfattede tilfælde af herpes simplex lungebetændelse urinvejsinfektion pharyngitis og herpes zoster.
I undersøgelsen oplevede JIA-I 45% af patienterne en infektion, mens han modtog Humira med eller uden samtidig MTX i de første 16 uger af behandlingen. De typer infektioner rapporteret hos HUMIRA-behandlede patienter svarede generelt til dem, der ofte blev set hos polyartikulære JIA-patienter, der ikke behandles med TNF-blokkeere. Efter påbegyndelse af behandling var de mest almindelige bivirkninger, der forekommer i denne patientpopulation behandlet med Humira, injektionsstedets smerte og reaktion på injektionsstedet (henholdsvis 19% og 16%). En mindre almindeligt rapporteret bivirkning hos patienter, der fik Humira, var granuloma annulare, hvilket ikke førte til seponering af Humira -behandling.
I de første 48 uger af behandlingen i undersøgelse blev JIA-I ikke-alvorlige overfølsomhedsreaktioner set hos ca. 6% af patienterne og inkluderede primært lokaliserede allergiske overfølsomhedsreaktioner og allergisk udslæt.
I undersøgelsen udviklede JIA-I 10% af patienterne, der blev behandlet med Humira, som havde negativ baseline-anti-dsDNA-antistoffer, positive titere efter 48 ugers behandling. Ingen patient udviklede kliniske tegn på autoimmunitet under det kliniske forsøg.
Cirka 15% af patienterne, der blev behandlet med HUMIRA, udviklede mild til moderate forhøjelser af kreatinphosphokinase (CPK) i undersøgelse JIA-I. Højde over 5 gange den øvre normale grænse blev observeret hos flere patienter. CPK -koncentrationer faldt eller vendte tilbage til det normale hos alle patienter. De fleste patienter var i stand til at fortsætte Humira uden afbrydelse.
I studiet blev Jia-II Humira undersøgt hos 32 patienter, der var 2 til <4 years of age or 4 years of age og older weighing <15 kg with polyarticular JIA. The safety profile for this patient population was similar to the safety profile seen in patients 4 til 17 år with polyarticular JIA.
I undersøgelsen oplevede JIA-II 78% af patienterne en infektion, mens han modtog Humira. Disse omfattede nasopharyngitis bronchitis øvre luftvejsinfektion otitis -medier og var for det meste milde til moderat i sværhedsgrad. Alvorlige infektioner blev observeret hos 9% af patienterne, der fik Humira i undersøgelsen og inkluderede tandkaries rotavirus gastroenteritis og varicella.
I undersøgelsen blev JIA-II ikke-alvorlige allergiske reaktioner observeret hos 6% af patienterne og inkluderede intermitterende urticaria og udslæt, som alle var milde i sværhedsgrad.
Psoriasis arthritis And Ankyloserende spondylitis Kliniske studier
Humira has been studied in 395 patients with psoriatic artritis (PsA) in two placebocontrolled trials og in an open label study og in 393 patients with ankylosing spondylitis (AS) in two placebo-controlled studies [see Kliniske studier ]. The safety profile for patients with PsA og AS treated with Humira 40 mg hver anden uge was similar to the safety profile seen in patients with RA Humira Studies RA-I through IV.
Crohns sygdomskliniske undersøgelser
Voksne
Sikkerhedsprofilen for HUMIRA hos 1478 voksne patienter med Crohns sygdom fra fire placebokontrollerede og to open-label-udvidelsesundersøgelser [se Kliniske studier ] svarede til sikkerhedsprofilen set hos patienter med RA.
Pædiatriske patienter 6 år til 17 år
Sikkerhedsprofilen for HUMIRA i 192 pædiatriske patienter fra en dobbeltblind undersøgelse (undersøgelse PCD-I) og en open-label-udvidelsesundersøgelse [se Kliniske studier ] svarede til sikkerhedsprofilen set hos voksne patienter med Crohns sygdom. I løbet af den 4-ugers åbne etiketinduktionsfase af undersøgelsen PCD-I var de mest almindelige bivirkninger, der forekommer i den pædiatriske population behandlet med Humira, injektionsstedssmerter og injektionsstedets reaktion (henholdsvis 6% og 5%).
I alt 67% af børnene oplevede en infektion, mens de modtog Humira i undersøgelse PCD-I. Disse omfattede øvre luftvejsinfektion og nasopharyngitis.
I alt 5% af børnene oplevede en alvorlig infektion, mens de modtog Humira i undersøgelse PCD-I. Disse omfattede viral infektionsindretningsrelateret sepsis (kateter) gastroenteritis H1N1 -influenza og formidlet histoplasmose.
I undersøgelsen blev PCD-I-allergiske reaktioner observeret hos 5% af børnene, som alle var ikke-seriøse og var primært lokaliserede reaktioner.
Ulcerøs colitis Kliniske studier
Voksne
Sikkerhedsprofilen for HUMIRA hos 1010 voksne patienter med ulcerøs colitis (UC) fra to placebokontrollerede studier og en open-label-udvidelsesundersøgelse [se Kliniske studier ] svarede til sikkerhedsprofilen set hos patienter med RA.
Pædiatriske patienter 5 år til 17 år
Sikkerhedsprofilen for HUMIRA hos 93 pædiatriske patienter med ulcerøs colitis fra en dobbeltblind undersøgelse og en open-label-udvidelsesundersøgelse [se Kliniske studier ] svarede til sikkerhedsprofilen set hos voksne patienter med ulcerøs colitis.
Plaque psoriasis Kliniske studier
Humira has been studied in 1696 subjects with plaque psoriasis (Ps) in placebo-controlled og open-label extension studies [see Kliniske studier ]. The safety profile for subjects with Ps treated with Humira was similar to the safety profile seen in subjects with RA with the following exceptions. In the placebo-controlled portions of the clinical trials in Ps subjects Humira-treated subjects had a higher incidence of arthralgia when comparød to controls (3% vs. 1%).
Hidradenitis suppurativ Kliniske studier
Humira has been studied in 727 subjects with hidradenitis suppurativa (HS) in three placebocontrolled studies og one open-label extension study [see Kliniske studier ]. The safety profile for subjects with HS treated with Humira weekly was consistent with the known safety profile of Humira.
Flare af HS defineret som ≥25% stigning fra baseline i abscesser og inflammatoriske knudepunktstællinger og med mindst 2 yderligere læsioner blev dokumenteret i 22 (22%) af de 100 forsøgspersoner, der blev trukket tilbage fra Humira -behandling efter den primære effektivitet i to undersøgelser.
Uveitis Kliniske studier
Humira has been studied in 464 adult patients with uveitis (UV) in placebo-controlled og open-label extension studies og in 90 pediatric patients with uveitis (Undersøg puv-i) [see Kliniske studier ]. Sikkerhedsprofilen for patienter med UV -behandlet med Humira svarede til sikkerhedsprofilen set hos patienter med RA.
Immunogenicitet
Som med alle terapeutiske proteiner er der potentiale for immunogenicitet. Påvisningen af antistofdannelse er meget afhængig af følsomheden og specificiteten af assayet. Derudover kan den observerede forekomst af antistof (inklusive neutraliserende antistof) positivitet i et assay påvirkes af adskillige faktorer, herunder assaymetodologi Prøvehåndteringstidspunkt for prøveopsamling Samtidig medicin og underliggende sygdom. Af disse grunde kan sammenligning af forekomsten af antistoffer i undersøgelsen beskrevet nedenfor med forekomsten af antistoffer i andre undersøgelser eller til andre adalimumab -produkter være vildledende.
Der er to assays, der er blevet anvendt til at måle anti-adalimumabantistoffer. Med ELISA -antistoffer til adalimumab kunne kun påvises, når serum adalimumab -koncentrationer var <2 mcg/mL. The ECL assay can detect anti-Adalimumab antibody titers independent of Adalimumab concentrations in the serum samples. The incidence of antiAdalimumab antibody (AAA) development in patients treated with Humira are presented in Table 2.
Tabel 2: Anti-adalimumab-antistofudvikling bestemt ved ELISA og ECL-assay hos patienter behandlet med Humira
| Indikationer | Undersøgelsesvarighed | Anti-adalimumab-antistofforekomst af ELISA (N/N) | Anti-adalimumab-antistofforekomst ved ECL-assay (N/N) | ||
| Hos alle patienter, der modtog adalimumab | Hos patienter med serum adalimumab -koncentrationer <2 mcg/mL | ||||
| Reumatoid arthritis a | 6 til 12 måneder | 5% (58/1062) | Ingen. | Na | |
| Juvenil idiopatisk arthritis (JIA) | 4 til 17 år b | 48 uger | 16% (27/171) | Ingen. | Na |
| 2 til 4 år eller ≥ 4 år og vejer <15 kg | 24 uger | 7% (1/15) c | Ingen. | Na | |
| Psoriasis arthritis d | 48 uger e | 13% (24/178) | Ingen. | Na | |
| Ankyloserende spondylitis | 24 uger | 9% (16/185) | Ingen. | Na | |
| Voksen Crohns sygdom | 56 uger | 3% (7/269) | 8% (7/86) | Na | |
| Pædiatrisk Crohns sygdom | 52 uger | 3% (6/182) | 10% (6/58) | Na | |
| Voksen ulcerøs colitis | 52 uger | 5% (19/360) | 21% (19/92) | Na | |
| Pædiatrisk ulcerøs colitis | 52 uger | 3% (3/100) | 13% (3/23) | 33% (33/100) i | |
| Plaque psoriasis f | Op til 52 uger g | 8% (77/920) | 21% (77/372) | Na | |
| Hidradenitis suppurativ | 36 uger | 7% (30/461) | 28% (58/207) h | 61% (272/445) j | |
| Ikke-infektiøs uveitis | 52 uger | 5% (12/249) | 21% (12/57) | 40% (99/249) k | |
| N: Antal patienter med anti-adalimumabantistof; NR: ikke rapporteret; NA: Ikke relevant (ikke udført) a Hos patienter, der fik samtidig methotrexat (MTX), var forekomsten af anti-adalimumab-antistof 1% sammenlignet med 12% med Humira monoterapi b Hos patienter, der fik samtidig MTX c Denne patient modtog samtidig MTX d Hos patienter, der modtog samtidig MTX e Motiver, der var tilmeldt efter at have afsluttet 2 tidligere undersøgelser på 24 uger eller 12 ugers behandlinger. f Hos plaque psoriasis -patienter, der var på Humira -monoterapi og derefter trukket tilbage fra behandlingen af antistoffer til adalimumab efter tilbagetrækning, svarede til den observerede sats før tilbagetrækning g En 12-ugers fase 2-undersøgelse og en 52-ugers fase 3-undersøgelse h Blandt forsøgspersoner i de 2 fase 3 -undersøgelser, der stoppede Humira -behandling i op til 24 uger, og i hvilke adalimumab -serumniveauer efterfølgende afviste til <2 mcg/mL (approximately 22% of total subjects studied) i Der blev ikke observeret nogen åbenbar sammenhæng mellem antistofudvikling og sikkerhed. Forbindelsen af antistofudvikling og effektivitetsresultat blev ikke vurderet på grund af et begrænset antal personer i hver behandlingsgruppe, der blev stratificeret af anti-adalimumab-antistof-titer. j Der blev ikke observeret nogen åbenbar sammenhæng mellem antistofudvikling og sikkerhed k Der blev ikke observeret nogen sammenhæng mellem antistofudvikling til sikkerheds- eller effektivitetsresultater |
Reumatoid arthritis And Psoriasis arthritis
Patienter i undersøgelser RA-I RA-II og RA-III blev testet på flere tidspunkter for antistoffer mod adalimumab ved hjælp af ELISA i løbet af 6- til 12month-perioden. Der blev ikke observeret nogen åbenbar sammenhæng mellem antistofudvikling til bivirkninger. Med monoterapipatienter, der modtager hver anden uges dosering, kan dosering udvikle antistoffer oftere end dem, der får ugentlig dosering. Hos patienter, der fik den anbefalede dosering på 40 mg hver anden uge som monoterapi, var ACR 20-responsen lavere blandt antistof-positive patienter end blandt antistofnegative patienter. Den langvarige immunogenicitet af Humira er ukendt.
Oplevelse af postmarketing
Følgende bivirkninger er blevet identificeret under anvendelse af Humira efter godkendelse. Fordi disse reaktioner rapporteres frivilligt fra en population af usikker størrelse, er det ikke altid muligt at pålideligt estimere deres frekvens eller etablere et årsagsforhold til Humira -eksponering.
Gastrointestinal disorders: Diverticulitis Store tarmperforeringer inklusive perforeringer forbundet med diverticulitis og appendiceal perforeringer forbundet med blindtarmsbetændelse pancreatitis
Generelle lidelser og administrationsstedets forhold: Pyrexia
Hepato-galdesygdomme: Leversvigt hepatitis
Immunsystemforstyrrelser: Sarkoidose
Neoplasmer godartet ondartet og uspecificeret (inklusive cyster og polypper): Merkelcellekarcinom (neuroendokrin karcinom i huden)
Nervesystemforstyrrelser: Demyeliniserende lidelser (f.eks. Optisk neuritis Guillain-Barré Syndrome) Cerebrovaskulær ulykke
Åndedrætsværn disorders: Interstitiel lungesygdom inklusive lungefibrose Lungeemboli
Hudreaktioner: Stevens Johnson syndrom Kutan vaskulitis erythema multiforme ny eller forværring af psoriasis (alle undertyper inklusive pustulær og palmoplantar) alopecia lichenoid hudreaktion
Vaskulære lidelser: Systemisk vaskulitis dyb venetrombose
Lægemiddelinteraktioner for Humira
Methotrexat
Humira has been studied in rheumatoid artritis (RA) patients taking concomitant methotrexate (MTX). Although MTX røduced the apparent Adalimumab clearance the data Gør ikke suggest the need for dose adjustment of either Humira or MTX [see Klinisk farmakologi ].
Biologiske produkter
I kliniske studier hos patienter med RA er der observeret en øget risiko for alvorlige infektioner med kombinationen af TNF -blokkeere med Anakinra eller abatacept uden ekstra fordel; Derfor anbefales brug af Humira med abatacept eller anakinra ikke hos patienter med RA [se ADVARSELS AND FORHOLDSREGLER ]. A higher rate of serious infections has also been observed in patients with RA treated with rituximab who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of Humira og other biologic products for the treatment of RA PsA AS CD UC Ps HS og UV. Concomitant administration of Humira with other biologic DMARDS (e.g. anakinra og abatacept) or other TNF blockers is ikke recommended based upon the possible increased risk for infections og other potential pharmacological interactions.
Live vacciner
Undgå brug af levende vacciner med Humira [se ADVARSELS AND FORHOLDSREGLER ].
Cytochrome P450 -underlag
Dannelsen af CYP450-enzymer kan undertrykkes ved forøgede koncentrationer af cytokiner (f.eks. TNFa IL-6) under kronisk inflammation. Det er muligt for et molekyle, der antagoniserer cytokinaktivitet, såsom adalimumab, at påvirke dannelsen af CYP450 -enzymer. Efter initiering eller seponering af HUMIRA hos patienter, der behandles med CYP450 -substrater med en smal terapeutisk indeksovervågning af effekten (f.eks. Warfarin) eller lægemiddelkoncentration (f.eks. Cyclosporin eller teofyllin) anbefales, og den individuelle dosis af lægemiddelproduktet kan justeres efter behov.
Advarsler for Humira
Inkluderet som en del af FORHOLDSREGLER afsnit.
Forholdsregler for Humira
Alvorlige infektioner
Patienter, der behandles med Humira, har en øget risiko for at udvikle alvorlige infektioner, der involverer forskellige organsystemer og steder, der kan føre til indlæggelse eller død. Opportunistiske infektioner på grund af bakterielle mycobakterielle invasive svampe virale parasitiske eller andre opportunistiske patogener, herunder aspergillosis blastomycosis candidiasis coccidioidomycosis histoplasmosis legionellosis listeriosis pneumocystosis og tuberculosis er blevet rapporteret med TNF -blockere. Patienter har ofte præsenteret for formidlet snarere end lokal sygdom.
Den samtidige anvendelse af en TNF -blokering og abatacept eller Anakinra var forbundet med en højere risiko for alvorlige infektioner hos patienter med reumatoid arthritis (RA); Derfor anbefales den samtidige brug af Humira og disse biologiske produkter ikke til behandling af patienter med RA [se ADVARSELS AND FORHOLDSREGLER og Lægemiddelinteraktioner ].
Behandling med Humira bør ikke initieres hos patienter med en aktiv infektion inklusive lokaliserede infektioner. Patienter 65 år og ældre patienter med co-morbide tilstande og/eller patienter, der tager samtidig immunsuppressiva (såsom kortikosteroider eller methotrexat) kan have større risiko for infektion. Overvej risikoen og fordelene ved behandling, inden behandlingen påbegyndes hos patienter:
- med kronisk eller tilbagevendende infektion;
- der er blevet udsat for tuberkulose;
- med en historie med en opportunistisk infektion;
- der har boet eller rejst i områder med endemisk tuberkulose eller endemiske mycoser, såsom histoplasmosis coccidioidomycosis eller blastomycosis; eller
- Med underliggende forhold, der kan disponere dem for infektion.
Tuberkulose
Tilfælde af reaktivering af tuberkulose og ny begyndelse af tuberkuloseinfektioner er rapporteret hos patienter, der får HUMIRA, herunder patienter, der tidligere har modtaget behandling for latent eller aktiv tuberkulose. Rapporter omfattede tilfælde af lunge og ekstrapulmonal (dvs. formidlet) tuberkulose. Evaluer patienter for tuberkulose -risikofaktorer og test for latent infektion inden indledningen af Humira og periodisk under terapi.
Behandling af latent tuberkuloseinfektion inden terapi med TNF -blokerende midler har vist sig at reducere risikoen for tuberkulose -reaktivering under terapi. Før man påbegyndes HUMIRA, skal du vurdere, om der er behov for behandling af latent tuberkulose; og overvej en induration på ≥ 5 mm en positiv tuberculin hudtestresultat, selv for patienter, der tidligere var vaccineret med Bacille Calmette-guerin (BCG).
Overvej anti-tuberkulosebehandling inden påbegyndelse af HUMIRA hos patienter med en tidligere historie med latent eller aktiv tuberkulose, i hvilken et passende behandlingsforløb ikke kan bekræftes, og for patienter med en negativ test for latent tuberkulose, men har risikofaktorer for tuberkuloseinfektion. På trods af profylaktisk behandling af tuberkulose er tilfælde af genaktiveret tuberkulose forekommet hos patienter behandlet med Humira. Konsultation med en læge med ekspertise i behandlingen af tuberkulose anbefales for at hjælpe i beslutningen om, hvorvidt at indlede antituberkulosebehandling er passende for en individuel patient.
Overvej stærkt tuberkulose i den differentielle diagnose hos patienter, der udvikler en ny infektion under Humira -behandling, især hos patienter, der tidligere eller for nylig har rejst til lande med en høj forekomst af tuberkulose, eller som har haft tæt kontakt med en person med aktiv tuberkulose.
Overvågning
Overvåg nøje patienter for udvikling af tegn og symptomer på infektion under og efter behandling med HUMIRA, herunder udviklingen af tuberkulose hos patienter, der testede negativt for latent tuberkuloseinfektion, før de påbegyndte terapi. Tests for latent tuberkuloseinfektion kan også være falsk negativ, mens den er på terapi med Humira.
Afbryd Humira, hvis en patient udvikler en alvorlig infektion eller sepsis. For a patient who develops a new infection during treatment with Humira closely monitor them perform a prompt og complete diagnostic workup appropriate for an immunocompromised patient og initiate appropriate antimicrobial therapy.
Invasive svampeinfektioner
Hvis patienter udvikler en alvorlig systemisk sygdom, og de bor eller rejser i regioner, hvor mycoser er endemisk, overvejer invasiv svampeinfektion i den differentielle diagnose. Antigen- og antistofprøvning til histoplasmose kan være negativ hos nogle patienter med aktiv infektion. Overvej passende empirisk antifungal terapi under hensyntagen til både risikoen for alvorlig svampeinfektion og risikoen for antifungal terapi, mens der udføres en diagnostisk oparbejdning. For at hjælpe med håndtering af sådanne patienter overvejer konsultation med en læge med ekspertise inden for diagnose og behandling af invasive svampeinfektioner.
Maligniteter
Overvej risikoen og fordelene ved TNF-blokkeringsbehandling inklusive HUMIRA, inden de initierer terapi hos patienter med en kendt malignitet end en succesfuld behandlet ikke-melanom hudkræft (NMSC), eller når man overvejer at fortsætte en TNF-blokering hos patienter, der udvikler en malignitet.
Maligniteter In Voksne
I de kontrollerede dele af kliniske forsøg med nogle TNF-blokkere, herunder Humira flere tilfælde af maligniteter, er der observeret blandt TNF-blokker-behandlede voksne patienter sammenlignet med kontrolbehandlede voksne patienter. Under de kontrollerede dele af 39 globale Humira -kliniske forsøg hos voksne patienter med reumatoid arthritis (RA) psoriasisartrit (PSA) ankyloserende spondylitis (As) Crohns sygdom (CD) ulcerøs colitis (UC) plaque psoriasis (PS) hidradenitis suppurativa (HS (UV) malignancies other than non-melanoma (basal cell and squamous cell) skin cancer were observed at a rate (95% confidence interval) of 0.7 (0.48 1.03) per 100 patient-years among 7973 HUMIRA-treated patients versus a rate of 0.7 (0.41 1.17) per 100 patient-years among 4848 control-treated patients (median duration of treatment of 4 months for HUMIRA-treated patients og 4 måneder for kontrolbehandlede patienter). I 52 globale kontrollerede og ukontrollerede kliniske forsøg med HUMIRA hos voksne patienter med RA PSA som CD UC PS HS og UV de mest observerede maligniteter andre end lymfom og NMSC var brystkolonprostatilunge og melanom. De maligniteter hos Humira-behandlede patienter i de kontrollerede og ukontrollerede dele af undersøgelserne var ens i type og antal som hvad der ville forventes i den generelle amerikanske befolkning i henhold til SEER-databasen (justeret for alderskøn og race) .1
I kontrollerede forsøg med andre TNF-blokkeere hos voksne patienter med højere risiko for maligniteter (dvs. patienter med KOL med en betydelig rygningshistorie og cyclophosphamid-behandlede patienter med Wegener's granulomatosis) skete en større del af maligniteter i TNF-blokkergruppen sammenlignet med kontrolgruppen.
Ikke-melanom hudkræft
Under de kontrollerede dele af 39 globale Humira-kliniske forsøg hos voksne patienter med RA PSA som CD UC PS HS og UV hastigheden (95% konfidensinterval) på NMSC var 0,8 (NULL,52 1,09) pr. 100 patientår blandt HUMIRA-behandlede patienter og 0,2 (NULL,10 0,59) pr. 100 patient-år blandt kontrol-behandlingspatienter. Undersøg alle patienter og især patienter med en medicinsk historie med forudgående langvarig immunsuppressiv terapi eller psoriasis -patienter med en historie med PUVA -behandling til tilstedeværelsen af NMSC før og under behandling med Humira.
Lymfom og leukæmi
I de kontrollerede dele af kliniske forsøg med alle TNF-blokkere hos voksne er der blevet observeret flere tilfælde af lymfom blandt TNF-blokker-behandlede patienter sammenlignet med kontrolbehandlede patienter. I de kontrollerede dele af 39 globale Humira-kliniske forsøg hos voksne patienter med RA PSA som CD UC PS HS og UV 2-lymfomer forekom blandt 7973 HUMIRA-behandlede patienter mod 1 blandt 4848 kontrolbehandlede patienter. I 52 globale kontrollerede og ukontrollerede kliniske forsøg med HUMIRA hos voksne patienter med RA PSA som CD UC PS HS og UV med en median varighed på ca. 0,7 år inklusive 24605 patienter og over 40215 patientår af Humira den observerede lymfomer var ca. 0,11 pr. 100 patient år. Dette er cirka 3 gange højere end forventet i den generelle amerikanske befolkning i henhold til SEER-databasen (justeret for alderskøn og race) .1 Hastigheder for lymfom i kliniske forsøg med HUMIRA kan ikke sammenlignes med hastighederne for lymfom i kliniske forsøg med andre TNF-blokkeere og kan ikke forudsige de hastigheder, der er observeret i en bredere patientpopulation. Patienter med RA og andre kroniske inflammatoriske sygdomme, især patienter med meget aktiv sygdom og/eller kronisk eksponering for immunsuppressive terapier, kan have en højere risiko (op til adskillige fold) end den generelle population for udvikling af lymfom, selv i fravær af TNF -blokkere. Tilfælde af akut og kronisk leukæmi efter markedsføring er rapporteret i forbindelse med TNF-blokkeringsbrug i RA og andre indikationer. Selv i fravær af TNF-blokkerterapipatienter med RA kan have en højere risiko (ca. 2 gange) end den generelle befolkning for udvikling af leukæmi.
Maligniteter In Pædiatriske patienter And Young Voksne
Maligniteter some fatal have been reported among children adolescents og young adults who received treatment with TNF-blockers (initiation of therapy ≤ 18 years of age) of which Humira is a member. Approximately half the cases were lymphomas including Hodgkin's og non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies og included rare malignancies usually associated with immunosuppression og malignancies that are ikke usually observed in children og adolescents. The malignancies occurrød after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing og are derived from a variety of sources including registries og spontaneous postmarketing reports.
Postmarkedstilfælde af hepatosplenisk T-celle-lymfom (HSTCL) En sjælden type T-celle-lymfom er rapporteret hos patienter behandlet med TNF-blokkeere inklusive HUMIRA. Disse tilfælde har haft et meget aggressivt sygdomsforløb og har været dødelig. Størstedelen af de rapporterede TNF -blokeringssager har forekommet hos patienter med Crohns sygdom eller ulcerøs colitis, og størstedelen var hos unge og unge voksne mænd. Næsten alle disse patienter havde modtaget behandling med immunsuppressiverne azathioprin eller 6-mercaptopurine (6â MP) samtidig med en TNF-blokkering ved eller før diagnosen. Det er usikkert, om forekomsten af HSTCL er relateret til brug af en TNF -blokering eller en TNF -blokering i kombination med disse andre immunsuppressiva. Den potentielle risiko ved kombinationen af azathioprin eller 6mercaptopurin og Humira bør overvejes omhyggeligt.
Overfølsomhedsreaktioner
Anafylaksi og angionurotisk ødem er rapporteret efter Humira -administration. Hvis der forekommer en anafylaktisk eller anden alvorlig allergisk reaktion, ophører med det samme administration af Humira og institutter passende terapi. I kliniske forsøg med Humira-overfølsomhedsreaktioner (f.eks. Udslæt anafylactoidreaktion er faste lægemiddelreaktion ikke-specificeret lægemiddelreaktion urticaria) blevet observeret.
Hepatitis B -virusreaktivering
Brug af TNF -blokkeere, herunder HUMIRA, kan øge risikoen for reaktivering af hepatitis B -virus (HBV) hos patienter, der er kroniske bærere af denne virus. I nogle tilfælde har HBV -reaktivering, der forekommer i forbindelse med TNF -blokeringsterapi, været dødelig. Størstedelen af disse rapporter har fundet sted hos patienter, der samtidig modtager andre medicin, der undertrykker immunsystemet, som også kan bidrage til HBV -reaktivering. Evaluer patienter, der er i fare for HBV -infektion for forudgående bevis for HBV -infektion, før de påbegyndte TNF -blokeringsterapi. Udøv forsigtighed ved ordination af TNF -blokkeere for patienter, der er identificeret som bærere af HBV. Tilstrækkelige data er ikke tilgængelige om sikkerheden eller effektiviteten af behandling af patienter, der er bærere af HBV med anti-viral terapi i forbindelse med TNF-blokeringsterapi for at forhindre HBV-reaktivering. For patienter, der er bærere af HBV og har brug for behandling med TNF -blokkeere, overvåger nøje sådanne patienter til kliniske og laboratorie -tegn på aktiv HBV -infektion gennem terapi og i flere måneder efter afslutning af terapi. Hos patienter, der udvikler HBV-reaktivering, stopper Humira og initierer effektiv anti-viral terapi med passende understøttende behandling. Sikkerheden ved genoptagelse af TNF -blokeringsterapi efter HBV -reaktivering er ikke kendt. Udvis derfor forsigtighed, når man overvejer genoptagelse af Humira -terapi i denne situation og overvåger patienter nøje.
Neurologiske reaktioner
Anvendelse af TNF-blokerende midler, herunder Humira, har været forbundet med sjældne tilfælde af ny begyndelse eller forværring af kliniske symptomer og/eller radiografisk bevis for centralnervesystem, der demyeliniserer sygdom, herunder multipel sklerose (MS) og optisk neuritis og perifer demyeliniserende sygdom, herunder guillain-tarrã © syndrom. Udøv forsigtighed ved at overveje brugen af HUMIRA hos patienter med forudgående eller nyligt begyndt centralt eller perifert nervesystem, der demonstrerer lidelser; Afbrydelse af Humira bør overvejes, hvis nogen af disse lidelser udvikler sig. Der er en kendt sammenhæng mellem mellemliggende uveitis og centrale demyeliniserende lidelser.
Hæmatologiske reaktioner
Sjældne rapporter om pancytopeni inklusive aplastisk anæmi er rapporteret med TNF -blokerende midler. Bivirkninger af det hæmatologiske system inklusive medicinsk signifikant cytopeni (f.eks. Thrombocytopenia leukopeni) er sjældent rapporteret med Humira. Årsagsforholdet mellem disse rapporter til Humira forbliver uklart. Rådgive alle patienter om at søge øjeblikkelig lægehjælp, hvis de udvikler tegn og symptomer, der tyder på bloddyscrasias eller infektion (f.eks. Vedvarende feber -blå mærker, der blødende blek), mens de er på Humira. Overvej seponering af HUMIRA -terapi hos patienter med bekræftede signifikante hæmatologiske abnormiteter.
Øget risiko for infektion, når det bruges sammen med Anakinra
Samtidig brug af Anakinra (en interleukin-1-antagonist) og en anden TNF-blokker var forbundet med en større andel af alvorlige infektioner og neutropeni og ingen ekstra fordel sammenlignet med TNF-blokkeren alene hos patienter med RA. Derfor anbefales kombinationen af Humira og Anakinra ikke [se Lægemiddelinteraktioner ].
Hjertesvigt
Tilfælde af forværring af kongestiv hjertesvigt (CHF) og nybegynder CHF er rapporteret med TNF -blokkeere. Tilfælde af forværring af CHF er også blevet observeret med Humira. Humira er ikke formelt undersøgt hos patienter med CHF; I kliniske forsøg med en anden TNF-blokering blev der imidlertid observeret en højere hastighed af alvorlige CHF-relaterede bivirkninger. Træk forsigtighed, når du bruger Humira hos patienter, der har hjertesvigt og overvåger dem omhyggeligt.
Autoimmunity
Behandling med Humira kan resultere i dannelse af autoantistoffer og sjældent i udviklingen af et lupuslignende syndrom. Hvis en patient udvikler symptomer, der tyder på et lupuslignende syndrom efter behandling med Humira, ophører behandlingen [se Bivirkninger ].
Immuniseringer
I et placebo-kontrolleret klinisk forsøg med patienter med RA blev der ikke påvist nogen forskel i antipneumokokk-antistofrespons mellem Humira og placebo-behandlingsgrupper, når de pneumokokk-polysaccharidvaccine og influenzavaccine blev administreret samtidig med Humira. Lignende andele af patienter udviklede beskyttelsesniveauer af anti-influenza-antistoffer mellem Humira og placebo-behandlingsgrupper; Imidlertid var titere samlet til influenzaantigener moderat lavere hos patienter, der fik Humira. Den kliniske betydning af dette er ukendt. Patienter på Humira kan modtage samtidige vaccinationer bortset fra levende vacciner. Ingen data er tilgængelige om den sekundære transmission af infektion af levende vacciner hos patienter, der får Humira.
Det anbefales, at pædiatriske patienter, hvis det er muligt, bringes ajour med alle immuniseringer i overensstemmelse med de nuværende immuniseringsretningslinjer, inden Humira -terapi indledte. Patienter på Humira kan modtage samtidige vaccinationer bortset fra levende vacciner.
Sikkerheden ved administration af levende eller levende dæmpede vacciner hos spædbørn udsat for Humira i utero er ukendt. Risici og fordele skal overvejes, inden de vaccineres (levende eller liveattenuerede) udsatte spædbørn [se Brug i specifikke populationer ].
Øget risiko for infektion, når det bruges sammen med abatacept
I kontrollerede forsøg var den samtidige administration af TNF-blokkere og abatacept forbundet med en større andel af alvorlige infektioner end brugen af en TNF-blokker alene; Kombinationsterapien sammenlignet med brugen af en TNF-blokker alene har ikke vist forbedret klinisk fordel i behandlingen af RA. Derfor anbefales kombinationen af abatacept med TNF-blokkere inklusive Humira ikke [se Lægemiddelinteraktioner ].
Oplysninger om patientrådgivning
Rådgive patienten eller plejeren om at læse den FDA-godkendte patientmærkning ( Medicin vejledning og Brug til brug ).
Infektioner
Informer patienter om, at Humira kan sænke deres immunsystems evne til at bekæmpe infektioner. Instruer patienter om vigtigheden af at kontakte deres læge, hvis de udvikler symptomer på infektion, herunder tuberkulose invasive svampeinfektioner og reaktivering af hepatitis B -virusinfektioner [se ADVARSELS AND FORHOLDSREGLER ].
Maligniteter
Rådgiver patienter om risikoen for maligniteter, mens de modtager Humira [se ADVARSELS AND FORHOLDSREGLER ]
Overfølsomhedsreaktioner
Rådgive patienter om at søge øjeblikkelig lægehjælp, hvis de oplever symptomer på alvorlige overfølsomhedsreaktioner. Rådgive latexfølsomme patienter om, at nålhætten på Humira 40 mg/0,8 ml pen og 40 mg/0,8 ml 20 mg/0,4 ml og 10 mg/0,2 ml præfyldt sprøjte kan indeholde naturlig gummi latex [se ADVARSELS AND FORHOLDSREGLER Hvor leveret / Opbevaring og håndtering ].
Andre Medical Conditions
Rådgiv patienter om at rapportere eventuelle tegn på nye eller forværrede medicinske tilstande, såsom kongestiv hjertesvigt neurologisk sygdom autoimmune lidelser eller cytopenier. Rådgiv patienter om at rapportere eventuelle symptomer, der tyder på en cytopeni, såsom blå blødning eller vedvarende feber [se ADVARSELS AND FORHOLDSREGLER ].
Instruktioner om injektionsteknik
Informer patienter om, at den første injektion skal udføres under opsyn af en kvalificeret sundhedspersonale. Hvis en patient eller en plejeperson skal administrere Humira instruere dem i injektionsteknikker og vurdere deres evne til at injicere subkutant for at sikre korrekt administration af Humira [se Brug til brug ].
For patienter, der vil bruge Humira -pen, fortæller dem, at de:
- Vil høre en høj â € ˜Klickâ € ™, når der trykkes på den blommefarvede aktivatorknap. Det høje klik betyder starten af injektionen.
- Skal fortsætte med at holde Humira -pennen mod deres klemte hævede hud, indtil al medicinen er injiceret. Dette kan tage op til 15 sekunder.
- Vil vide, at injektionen er afsluttet, når den gule markør fuldt ud vises i vinduesvisningen og holder op med at bevæge sig.
Instruer patienter om at bortskaffe deres brugte nåle og sprøjter eller brugte pen i en FDA-klaret skarp bortskaffelsesbeholder umiddelbart efter brug. Instruer patienter om ikke at bortskaffe løse nåle og sprøjter eller pen i deres husholdningspass. Instruer patienter om, at hvis de ikke har en FDA-klaret skarp bortskaffelsesbeholder, kan de bruge en husholdningscontainer, der er lavet af en kraftig plastik, kan lukkes med en tæt passende og punkteringsbestandig låg uden at skarpe er i stand til at komme ud oprejst og stabilt under brug af lækagebestandigt og korrekt mærket for at advare om farligt affald inde i beholderen.
Instruer patienter om, at når deres skarpe bortskaffelsescontainer næsten er fuld, skal de følge deres samfundsretningslinjer for den rigtige måde at bortskaffe deres skarpe bortskaffelsescontainer. Instruer patienter om, at der kan være statslige eller lokale love vedrørende bortskaffelse af brugte nåle og sprøjter. Henvis patienter til FDAs websted på https://www.fda.gov/safesharpsdisposal for mere information om sikker Sharps bortskaffelse og for specifik information om Sharps -bortskaffelse i staten, som de bor i.
Instruer patienter om ikke at bortskaffe deres brugte Sharps -bortskaffelsescontainer i deres husholdningspasselinjer, medmindre deres retningslinjer for samfundet tillader dette. Instruer patienter om ikke at genbruge deres brugte skarpe bortskaffelsescontainer.
Ikke -klinisk toksikologi
Karcinogenese mutagenese nedskrivning af fertilitet
Langvarige dyreforsøg af Humira er ikke blevet udført for at evaluere det kræftfremkaldende potentiale eller dets virkning på fertiliteten.
Brug i specifikke populationer
Graviditet
Risikooversigt
Tilgængelige undersøgelser med brug af adalimumab under graviditet opretter ikke pålideligt en sammenhæng mellem adalimumab og større fødselsdefekter. Kliniske data er tilgængelige fra Organisation of Teratology Information Specialists (OTIS)/Mothertobaby Humira Graviditetsregister hos gravide kvinder med reumatoid arthritis (RA) eller Crohns sygdom (CD). Registreringsresultater viste en sats på 10% for større fødselsdefekter med første trimesterbrug af adalimumab hos gravide kvinder med RA eller CD og en sats på 7,5% for større fødselsdefekter i den dyrebare sammenligningskohort. Manglen på mønster af større fødselsdefekter er betryggende, og forskelle mellem eksponeringsgrupper kan have påvirket forekomsten af fødselsdefekter (se Data ).
Adalimumab overføres aktivt over morkagen i tredje trimester af graviditeten og kan påvirke immunresponsen i det in-utero udsatte spædbarn (se Kliniske overvejelser ). In an embryo-fetal perinatal development study conducted in cynomolgus monkeys no fetal harm or malformations were observed with intravenous administration of Adalimumab during organogenesis og later in gestation at doses that produced exposures up to approximately 373 times the maximum recommended human dose (MRHD) of 40 mg subcutaneous without methotrexate (see Data ).
Den estimerede baggrundsrisiko for store fødselsdefekter og spontanabort for de angivne populationer er ukendt. Alle graviditeter har en baggrundsrisiko for tab af fødselsdefekt eller andre bivirkninger. I den amerikanske generelle befolkning er den estimerede baggrundsrisiko for større fødselsdefekter og spontanabort i klinisk anerkendte graviditeter henholdsvis 2-4% og 15-20%.
Kliniske overvejelser
Sygdomsassocieret moderlig og embryo/føtal risiko
Publicerede data antyder, at risikoen for ugunstige graviditetsresultater hos kvinder med RA eller inflammatorisk tarmsygdom (IBD) er forbundet med øget sygdomsaktivitet. Bivirkning af graviditet inkluderer for tidlig levering (før 37 ugers drægtighed) lav fødselsvægt (mindre end 2500 g) spædbørn og små til svangerskabsalder ved fødslen.
Føtal/neonatal bivirkninger
Monoklonale antistoffer transporteres i stigende Data ). Risks og benefits should be considerød prior to administering live or live-attenuated vaccines to infants exposed to Humira i utero [see Brug i specifikke populationer ].
Data
Menneskelige data
Et potentielt eksponeringsregister for graviditetseksponering for graviditet udført af Otis/Mothertobaby i USA og Canada mellem 2004 og 2016 sammenlignede risikoen for store fødselsdefekter hos levende fødte spædbørn på 221 kvinder (69 RA 152 CD) behandlet med Adalimumab i første trimester og 106 kvinder (74 RA 32 cd) ikke behandlet med Adalimumab.
Andelen af større fødselsdefekter blandt levende fødte spædbørn i de adalimumab-behandlede og ubehandlede kohorter var henholdsvis 10% (NULL,7% RA 10,5% CD) og 7,5% (NULL,8% RA 9,4% CD). Manglen på mønster af større fødselsdefekter er betryggende, og forskelle mellem eksponeringsgrupper kan have påvirket forekomsten af fødselsdefekter. Denne undersøgelse kan ikke pålideligt fastlægge, om der er en sammenhæng mellem adalimumab og større fødselsdefekter på grund af metodologiske begrænsninger i registreringsdatabasen, herunder lille prøvestørrelse den frivillige karakter af undersøgelsen og det ikke-randomiserede design.
I en uafhængig klinisk undersøgelse udført i ti gravide kvinder med IBD blev behandlet med Humira Adalimumab -koncentrationer målt i moderserum såvel som i ledningsblod (n = 10) og spædbarnsserum (n = 8) på fødslen. Den sidste dosis af Humira blev givet mellem 1 og 56 dage før levering. Adalimumab-koncentrationer var 0,16-19,7 μg/ml i ledningsblod 4,28-17,7 μg/ml i spædbarnsserum og 0-16,1 μg/ml i moderlig serum. I alle undtagen et tilfælde var snorblodkoncentrationen af adalimumab højere end moderens serumkoncentration, der antyder, at adalimumab aktivt krydser placenta. Derudover havde et spædbarn serumkoncentrationer ved hver af følgende: 6 uger (NULL,94 μg/ml) 7 uger (NULL,31 μg/ml) 8 uger (NULL,93 μg/ml) og 11 uger (NULL,53 μg/ml), hvilket antyder, at adalimumab kan påvises i serum af spædbørn, der blev udsat i utero i mindst 3 måneder fra fødslen.
Dyredata
I en embryo-føtal perinatal udviklingsundersøgelse modtog gravide cynomolgus-aber adalimumab fra drægtighed dage 20 til 97 i doser, der producerede eksponeringer op til 373 gange, der opnåede med MRHD uden methotrexat (på AUC-basis med modernal IV-doser op til 100 mg/kg/uge). Adalimumab fremkaldte ikke skade på fostre eller misdannelser.
Amning
Risikooversigt
Begrænsede data fra sagsrapporter i den offentliggjorte litteratur beskriver tilstedeværelsen af adalimumab i human mælk ved spædbarnsdoser på 0,1% til 1% af moderens serumkoncentration. Publicerede data antyder, at den systemiske eksponering for et ammet spædbarn forventes at være lav, fordi adalimumab er et stort molekyle og nedbrydes i mave -tarmkanalen. Virkningerne af lokal eksponering i mave -tarmkanalen er imidlertid ukendte. Der er ingen rapporter om bivirkninger af adalimumab på det ammede spædbarn og ingen effekter på mælkeproduktionen. De udviklingsmæssige og sundhedsmæssige fordele ved amning bør overvejes sammen med moders kliniske behov for Humira og eventuelle bivirkninger på det ammede barn fra Humira eller fra den underliggende moderlige tilstand.
Pædiatrisk brug
Humiras sikkerhed og effektivitet er blevet etableret for:
- Reduktion af tegn og symptomer på moderat til alvorligt aktiv polyartikulær JIA hos pædiatriske patienter 2 år og ældre.
- Behandlingen af moderat til alvorligt aktiv Crohns sygdom hos pædiatriske patienter 6 år og ældre.
- Behandlingen af moderat til alvorligt aktiv ulcerøs colitis hos pædiatriske patienter 5 år og ældre.
- Behandlingen af moderat til svær hidradenitis suppurativa hos patienter 12 år og ældre.
- Behandlingen af ikke-infektiøs mellemliggende posterior og panuveitis hos pædiatriske patienter 2 år og ældre.
På grund af dens inhibering af TNFa-humira, der blev administreret under graviditet, kunne det påvirke immunresponsen i den utero-eksponerede nyfødte og spædbarn. Data fra otte spædbørn udsat for Humira i utero antyder, at adalimumab krydser placenta [se Brug i specifikke populationer ]. The clinical significance of elevated Adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks og benefits should be considerød prior to vaccinating (live or live-attenuated) exposed infants.
Tilfælde af lymfom efter markedsføring af lymfom inklusive hepatosplenisk T-celle-lymfom og andre maligniteter, som nogle dødelige er rapporteret blandt børn, unge og unge voksne, der modtog behandling med TNF-blokkere, inklusive Humira [se ADVARSELS AND FORHOLDSREGLER ].
Juvenil idiopatisk arthritis
I studiet blev Jia-I Humira vist at reducere tegn og symptomer på aktiv polyartikulær JIA hos patienter 4 til 17 år [se Kliniske studier ]. In Undersøg Jia-iI the safety profile for patients 2 to <4 years of age was similar to the safety profile for patients 4 til 17 år with polyarticular JIA [see Bivirkninger ]. Humira has ikke been studied in patients with polyarticular JIA less than 2 years of age or in patients with a weight below 10 kg.
Sikkerheden af Humira hos patienter i de polyartikulære JIA -forsøg svarede generelt til den, der blev observeret hos voksne med visse undtagelser [se Bivirkninger ].
Sikkerheden og effektiviteten af Humira er ikke blevet etableret hos pædiatriske patienter med JIA mindre end 2 år.
Pædiatrisk Crohns sygdom
Humiras sikkerhed og effektivitet til behandling af moderat til alvorligt aktiv Crohns sygdom er blevet etableret hos pædiatriske patienter 6 år og ældre. Brug af Humira til denne indikation understøttes af bevis fra tilstrækkelige og velkontrollerede studier hos voksne med yderligere data fra en randomiseret dobbeltblind 52-ugers klinisk undersøgelse af to dosiskoncentrationer af Humira i 192 pædiatriske patienter (6 år til 17 år) [se Bivirkninger Klinisk farmakologi Kliniske studier ]. The adverse reaction profile in patients 6 years to 17 years of age was similar to adults.
Sikkerheden og effektiviteten af Humira er ikke blevet etableret hos pædiatriske patienter med Crohns sygdom mindre end 6 år.
Pædiatrisk ulcerøs colitis
Sikkerheden og effektiviteten af HUMIRA til behandling af moderat til alvorligt aktiv ulcerøs colitis er blevet etableret hos pædiatriske patienter 5 år og ældre. Brug af Humira til denne indikation understøttes af bevis fra tilstrækkelige og velkontrollerede studier hos voksne med yderligere data fra en randomiseret dobbeltblind 52-ugers klinisk undersøgelse af to dosiskoncentrationer af Humira hos 93 pædiatriske patienter (5 år til 17 år) [se Bivirkninger Klinisk farmakologi Kliniske studier ]. The adverse reaction profile in patients 5 years to 17 years of age was similar to adults.
Effektiviteten af HUMIRA er ikke blevet fastlagt hos patienter, der har mistet responsen eller var intolerante over for TNF -blokkeere.
Sikkerheden og effektiviteten af Humira er ikke blevet etableret hos pædiatriske patienter med ulcerøs colitis mindre end 5 år.
Pædiatrisk uveitis
Humiras sikkerhed og effektivitet til behandling af ikke-infektiøs uveitis er blevet fastlagt hos pædiatriske patienter 2 år og ældre. Brugen af Humira understøttes af bevis fra tilstrækkelige og velkontrollerede undersøgelser af Humira hos voksne og en 2: 1 randomiseret kontrolleret klinisk undersøgelse hos 90 pædiatriske patienter [se Kliniske studier ]. The safety og effectiveness of Humira have ikke been established in pediatric patients with uveitis less than 2 years of age.
Hidradenitis suppurativ
Brug af Humira hos pædiatriske patienter, der er 12 år og ældre for HS, understøttes af bevis fra tilstrækkelige og velkontrollerede undersøgelser af Humira hos voksne HS-patienter. Yderligere population af farmakokinetisk modellering og simulering forudsagde, at vægtbaseret dosering af Humira hos pædiatriske patienter 12 år og ældre kan give generelt lignende eksponering for voksne HS-patienter. Forløbet af HS er tilstrækkeligt ens hos voksne og unge patienter til at tillade ekstrapolering af data fra voksne til unge patienter. Den anbefalede dosering hos pædiatriske patienter, der er 12 år eller ældre, er baseret på kropsvægt [se Dosering og administration Klinisk farmakologi og Kliniske studier ].
Sikkerheden og effektiviteten af Humira er ikke blevet etableret hos patienter under 12 år med HS.
Geriatrisk brug
I alt 519 RA-patienter 65 år og ældre inklusive 107 patienter 75 år og ældre modtog Humira i kliniske studier RA-I gennem IV. Der blev ikke observeret nogen samlet forskel i effektivitet mellem disse patienter og yngre patienter. Hyppigheden af alvorlig infektion og malignitet blandt Humira -behandlede patienter 65 år og ældre var højere end for dem, der var mindre end 65 år. Overvej fordelene og risiciene ved Humira hos patienter 65 år og ældre. Hos patienter, der er behandlet med Humira, overvåger nøje for udviklingen af infektion eller malignitet [se ADVARSELS AND FORHOLDSREGLER ].
Overdoseringsoplysninger til Humira
Doser op til 10 mg/kg er blevet administreret til patienter i kliniske forsøg uden bevis for dosisbegrænsende toksiciteter. I tilfælde af overdosering anbefales det, at patienten overvåges for tegn eller symptomer på bivirkninger eller virkninger og passende symptomatisk behandling, der straks blev indført.
Kontraindikationer for Humira
Ingen.
Klinisk farmakologi for Humira
Handlingsmekanisme
Adalimumab binder specifikt til TNF-alfa og blokerer dens interaktion med p55- og p75-celleoverflade-TNF-receptorerne. Adalimumab lyser også overflade TNF -udtrykkende celler in vitro i nærvær af komplement. Adalimumab binder ikke eller inaktiverer lymfotoksin (TNF-beta). TNF er et naturligt forekommende cytokin, der er involveret i normale inflammatoriske og immunresponser. Forhøjede koncentrationer af TNF findes i synovialvæsken hos patienter med RA JIA PSA og AS og spiller en vigtig rolle i både den patologiske betændelse og den leddestruktion, der er kendetegnende for disse sygdomme. Forøgede koncentrationer af TNF findes også i psoriasis -plaques. I PS -behandling med Humira kan reducere den epidermale tykkelse og infiltration af inflammatoriske celler. Forholdet mellem disse farmakodynamiske aktiviteter og mekanismen (er), hvormed Humira udøver sine kliniske effekter er ukendt.
Adalimumab modulerer også biologiske responser, der induceres eller reguleres af TNF, herunder ændringer i koncentrationerne af adhæsionsmolekyler, der er ansvarlige for leukocytmigration (ELAM-1 VCAM-1 og ICAM-1 med en IC50 på 1-2 x 10-10m).
Farmakodynamik
Efter behandling med HUMIRA et fald i koncentrationer af akutte fase-reaktanter af inflammation (C-reaktivt protein [CRP] og erythrocytsedimentationshastighed [ESR]) og serumcytokiner (IL-6) blev observeret sammenlignet med baseline hos patienter med rheumatoid arthritis. Et fald i CRP -koncentrationer blev også observeret hos patienter med Crohns sygdoms ulcerøs colitis og hidradenitis suppurativa. Serumkoncentrationer af matrixmetalloproteinaser (MMP-1 og MMP-3), der producerer vævsombygning, der er ansvarlig for ødelæggelse af brusk, blev også reduceret efter Humira-administration.
For pædiatriske patienter 5 år til 17 år med ulcerøs colitis er den anbefalede dosering af Humira baseret på modelleret dosis/eksponeringseffektivitetsforhold og farmakokinetiske data. Der er ingen forventede klinisk relevante forskelle i effektivitet mellem den studerede højere dosering, der administreres i det kliniske forsøg (uger 0 til 52 i undersøgelse PUC-I) [se Kliniske studier ] og den anbefalede dosering [se Dosering og administration ].
Farmakokinetik
Farmakokinetikken af adalimumab var lineær over dosisområdet fra 0,5 til 10 mg/kg efter administration af en enkelt intravenøs dosis (Humira er ikke godkendt til intravenøs anvendelse). Efter 20 40 og 80 mg hver anden uge og hver uge subkutan administration Adalimumab gennemsnitlige serumtrugkoncentrationer i stabil tilstand steg omtrent proportionalt med dosis hos RA -patienter. Den gennemsnitlige terminale halveringstid var cirka 2 uger fra 10 til 20 dage på tværs af undersøgelser. Sunde forsøgspersoner og patienter med RA viste lignende adalimumab farmakokinetik.
Adalimumab -eksponering hos patienter behandlet med 80 mg hver anden uge vurderes at være sammenlignelig med den hos patienter, der blev behandlet med 40 mg hver uge.
Absorption
Den gennemsnitlige absolutte biotilgængelighed af adalimumab efter en enkelt 40 mg subkutan dosis var 64%. Den gennemsnitlige tid til at nå den maksimale koncentration var 5,5 dage (131 ± 56 timer), og den maksimale serumkoncentration var 4,7 ± 1,6 mcg/ml hos raske forsøgspersoner efter en enkelt 40 mg subkutan administration af Humira.
Fordeling
Distributionsvolumen (VSS) varierede fra 4,7 til 6,0 L efter intravenøs administration af doser, der spænder fra 0,25 til 10 mg/kg hos RA -patienter.
Eliminering
Den enkelte dosis farmakokinetik af adalimumab hos RA -patienter blev bestemt i flere undersøgelser med intravenøse doser, der spænder fra 0,25 til 10 mg/kg. Den systemiske clearance af adalimumab er ca. 12 ml/t. I langtidsundersøgelser med dosering mere end to år var der ingen bevis for ændringer i clearance over tid hos RA-patienter.
Patientpopulation
Reumatoid arthritis And Ankyloserende spondylitis
Hos patienter, der fik 40 mg Humira hver anden uge, var adalimumab gennemsnitlig steady-state trug-koncentrationer ca. 5 mcg/ml og 8 til 9 mcg/ml uden og med MTX samtidig behandling. Adalimumab -koncentrationer i synovialvæsken fra fem reumatoid arthritispatienter varierede fra 31 til 96% af dem i serum. Farmakokinetikken for adalimumab hos patienter med AS svarede til dem hos patienter med RA.
Psoriasis arthritis
Hos patienter, der fik 40 mg hver anden uge, var adalimumab-gennemsnitlig steady-state trug-koncentrationer 6 til 10 mcg/ml og 8,5 til 12 mcg/ml uden og med MTX samtidig behandling.
Plaque psoriasis
Adalimumab gennemsnitlig steady-state trug-koncentration var ca. 5 til 6 mcg/ml under Humira 40 mg hver anden uge-behandling.
Voksen uveitis
Adalimumab gennemsnitlig stabil koncentration var ca. 8 til 10 mcg/ml under Humira 40 mg hver anden ugebehandling.
Voksen hidradenitis suppurativ
Adalimumab-trugkoncentrationer var ca. 7 til 8 mcg/ml i henholdsvis uge 2 og uge 4 efter at have modtaget 160 mg i uge 0 efterfulgt af 80 mg i uge 2. gennemsnitlig steady-state trug-koncentration i uge 12 til uge 36 var ca. 7 til 11 mcg/ml under Humira 40 mg behandling af ugen.
Voksen Crohns sygdom
Adalimumab middeltrugkoncentrationer var ca. 12 mcg/ml i uge 2 og uge 4 efter at have modtaget 160 mg i uge 0 efterfulgt af 80 mg i uge 2. middelstats-state trugkoncentrationer var 7 mcg/ml i uge 24 og uge 56 under Humira 40 mg hver anden ugebehandling.
Voksen ulcerøs colitis
Adalimumab gennemsnitlige trugkoncentrationer var ca. 12 mcg/ml i uge 2 og uge 4 efter at have modtaget 160 mg i uge 0 efterfulgt af 80 mg i uge 2. gennemsnitlig steady-state trug-koncentrationer var ca. 8 mcg/ml og 15 mcg/ml i uge 52 efter at have modtaget en dosis af Humira 40 mg hver anden uge og 40 mg hver uge henholdsvis ugen.
Anti-lægemiddelantistofeffekter på farmakokinetikken
Reumatoid arthritis
En tendens mod højere tilsyneladende clearance af adalimumab i nærvær af anti-adalimumab-antistoffer blev identificeret.
Pædiatrisk ulcerøs colitis
Antistoffer mod adalimumab ved ECL -assay var forbundet med reduceret serum adalimumab -koncentrationer hos pædiatriske patienter med moderat til alvorligt aktiv ulcerøs colitis.
Hidradenitis suppurativ
Hos personer med moderat til svære HS -antistoffer mod adalimumab var der forbundet med reducerede serum adalimumab -koncentrationer. Generelt er omfanget af reduktion i serum adalimumab -koncentrationer større med stigende titere af antistoffer mod adalimumab.
Specifikke populationer
Geriatriske patienter
En lavere clearance med stigende alder blev observeret hos patienter med RA i alderen 40 til> 75 år.
Pædiatriske patienter
Juvenil idiopatisk arthritis
- 4 år til 17 år: Adalimumab-gennemsnittet af steady-state trug-koncentrationer var 6,8 mcg/ml og 10,9 mcg/ml hos patienter, der vejer <30 kg receiving 20 mg Humira subcutaneously every other week as moikkeherapy or with concomitant MTX respectively. The Adalimumab mean steady-state trough concentrations were 6.6 mcg/mL og 8.1 mcg/mL in patients weighing ≥30 kg receiving 40 mg Humira subcutaneously every other week as moikkeherapy or with MTX concomitant treatment respectively.
- 2 år til <4 years of age or 4 years of age og older weighing <15 kg: The Adalimumab mean steady-state trough Adalimumab concentrations were 6.0 mcg/mL og 7.9 mcg/mL in patients receiving Humira subcutaneously every other week as moikkeherapy or with MTX concomitant treatment respectively.
Pædiatrisk hidradenitis suppurativa
Adalimumab -koncentrationer hos unge patienter med HS, der modtager de anbefalede doseringsregimer, forventes at svare til dem, der er observeret hos voksne personer med HS baseret på population af farmakokinetisk modellering og simulering.
Pædiatrisk Crohns sygdom
Adalimumab gennemsnit â ± SD -koncentrationer var 15,7 ± 6,5 mcg/ml i uge 4 efter 160 mg i uge 0 og 80 mg i uge 2 og 10,5 ± 6,0 mcg/ml i uge 52 efter 40 mg hver anden uge dosering hos patienter, der vejer ≥ 40 kg. Adalimumab gennemsnit â ± SD -koncentrationer var 10,6 ± 6,1 mcg/ml ved uge 4 efter dosering 80 mg ved uge 0 og 40 mg i uge 2 og 6,9 ± 3,6 mcg/ml i uge 52 efter 20 mg hver anden uge dosering hos patienter, der vejer <40 kg.
Pædiatrisk ulcerøs colitis
Adalimumab gennemsnitlig steady-state trug-koncentration var 5,0 ± 3,3 mcg/ml i uge 52 efter subkutan administration på 0,6 mg/kg (maksimalt 40 mg) hver anden uge i pædiatriske UC-patienter 5 år til 17 år gammel. Hos patienter, der modtog 0,6 mg/kg (maksimalt 40 mg) hver uge, var den gennemsnitlige steady-state trug-koncentration 15,7 ± 5,6 mcg/ml i uge 52 hos pædiatriske UC-patienter 5 år til 17 år.
Mandlige og kvindelige patienter
Der blev ikke observeret nogen kønsrelaterede farmakokinetiske forskelle efter korrektion for en patients kropsvægt. Sunde forsøgspersoner og patienter med reumatoid arthritis udviste lignende adalimumab farmakokinetik.
Patienter med nedsat nyre- eller leverfunktion
Ingen farmakokinetiske data er tilgængelige hos patienter med lever- eller nyrefunktion.
Rheumatoid faktor eller CRP -koncentrationer
Mindre stigninger i tilsyneladende clearance blev forudsagt hos RA -patienter, der fik doser lavere end den anbefalede dosis og hos RA -patienter med høj reumatoid faktor eller CRP -koncentration. Disse stigninger er sandsynligvis ikke klinisk vigtige.
Lægemiddelinteraktionsundersøgelser
Methotrexat
MTX reducerede Adalimumab tilsyneladende godkendelse efter enkelt og flere dosering med henholdsvis 29% og 44% hos patienter med RA [se Lægemiddelinteraktioner ].
Kliniske studier
Reumatoid arthritis
Effektiviteten og sikkerheden af Humira blev vurderet i fem randomiserede dobbeltblinde undersøgelser hos patienter ≥18 år med aktiv reumatoid arthritis (RA) diagnosticeret i henhold til American College of Rheumatology (ACR) -kriterier. Patienter havde mindst 6 hævede og 9 ømme led. Humira blev administreret subkutant i kombination med methotrexat (MTX) (NULL,5 til 25 mg undersøgelser RA-I RA-III og RA-V) eller som monoterapi (undersøgelser RA-II).
Undersøgelse RA-I evaluerede 271 patienter, der havde mislykket terapi med mindst en, men ikke mere end fire DMARD'er og havde utilstrækkelig respons på MTX. Doser på 20 40 eller 80 mg Humira eller placebo blev givet hver anden uge i 24 uger.
Undersøgelse RA-II evaluerede 544 patienter, der havde mislykket terapi med mindst en DMARD. Doser af placebo 20 eller 40 mg Humira blev givet som monoterapi hver anden uge eller ugentligt i 26 uger.
Undersøgelse RA-III evaluerede 619 patienter, der havde en utilstrækkelig respons på MTX. Patienter modtog placebo 40 mg Humira hver anden uge med placebo -injektioner på alternative uger eller 20 mg Humira ugentligt i op til 52 uger. Undersøgelse RA-III havde et yderligere primært slutpunkt ved 52 ugers inhibering af sygdomsprogression (som detekteret ved røntgenresultater). Efter afslutningen af de første 52 uger, 457 patienter, der var indskrevet i en open-label-forlængelsesfase, hvor 40 mg Humira blev administreret hver anden uge i op til 5 år.
Undersøgelse RA-IV vurderede sikkerhed hos 636 patienter, der enten var DMARD-naive eller fik lov til at forblive på deres allerede eksisterende reumatologiske terapi, forudsat at terapi var stabil i mindst 28 dage. Patienter blev randomiseret til 40 mg Humira eller placebo hver anden uge i 24 uger.
Undersøgelse RA-V evaluerede 799 patienter med moderat til alvorligt aktiv RA på mindre end 3 års varighed, der var ≥18 år gamle og MTX Naã¯ve. Patienter blev randomiseret til at modtage enten MTX (optimeret til 20 mg/uge for uge 8) Humira 40 mg hver anden uge eller HUMIRA/MTX -kombinationsterapi i 104 uger. Patienter blev evalueret for tegn og symptomer og for radiografisk progression af ledskader. Median sygdomsvarighed blandt patienter, der var indskrevet i undersøgelsen, var 5 måneder. Den median MTX -dosis opnået var 20 mg.
Klinisk respons
Procenten af Humira -behandlede patienter, der opnåede ACR 20 50 og 70 responser i undersøgelser RAII og III, er vist i tabel 3.
Tabel 3: ACR-svar i studier RA-II og RA-III (procent af patienterne)
| Svar | Undersøg RA-II monoterapi (26 uger) | Undersøg RA-III methotrexatkombination (24 og 52 uger) | |||
| Placebo N = 110 | Humira 40 mg hver anden uge N = 113 | Humira 40 mg weekly N = 103 | Placebo/ MTX N = 200 | Humira/ MTX 40 mg hver anden uge N = 207 | |
| ACR20 | |||||
| Måned 6 | 19% | 46%* | 53%* | 30% | 63%* |
| Måned 12 | Na | Na | Na | 24% | 59%* |
| ACR50 | |||||
| Måned 6 | 8% | 22%* | 35%* | 10% | 39%* |
| Måned 12 | Na | Na | Na | 10% | 42%* |
| ACR70 | |||||
| Måned 6 | 2% | 12%* | 18%* | 3% | 21%* |
| Måned 12 | Na | Na | Na | 5% | 23%* |
| * s <0.01 Humira vs. placebo |
Resultaterne af undersøgelse RA-I svarede til undersøgelse RA-III; Patienter, der modtog Humira 40 mg hver anden uge i undersøgelse RA-I opnåede også ACR 20 50 og 70 svarprocent på henholdsvis 65% 52% og 24% sammenlignet med placebo-svar på henholdsvis 13% 7% og 3% efter 6 måneder (P <0.01).
Resultaterne af komponenterne i ACR-responskriterierne for undersøgelser RA-II og RA-III er vist i tabel 4. ACR-responsrater og forbedring af alle komponenter i ACR-respons blev opretholdt til uge 104. I løbet af de 2 år i undersøgelse af RA-III 20% af Humira-patienter, der fik 40 mg hver anden uge, opnåede en større klinisk respons defineret som vedligeholdelse af en ACR 70-respons over en 6-måned-periode. ACR-svar blev opretholdt i lignende proportioner af patienter i op til 5 år med kontinuerlig Humira-behandling i den åbne del af undersøgelsen RA-III.
Tabel 4: Komponenter i ACR-respons i studier RA-II og RA-III
| Parameter (median) | Undersøg RA-II | Undersøg RA-III | ||||||
| Placebo N = 110 | Humira a N = 113 | Placebo/MTX N = 200 | Humira a /MTX N = 207 | |||||
| Baseline | WK 26 | Baseline | WK 26 | Baseline | WK 24 | Baseline | WK 24 | |
| Antal ømme led (0-68) | 35 | 26 | 31 | 16* | 26 | 15 | 24 | 8* |
| Antal hævede led (0-66) | 19 | 16 | 18 | 10* | 17 | 11 | 18 | 5* |
| Læge Global vurdering b | 7.0 | 6.1 | 6.6 | 3.7* | 6.3 | 3.5 | 6.5 | 2.0* |
| Patientens globale vurdering b | 7.5 | 6.3 | 7.5 | 4.5* | 5.4 | 3.9 | 5.2 | 2.0* |
| Smerte b | 7.3 | 6.1 | 7.3 | 4.1* | 6.0 | 3.8 | 5.8 | 2.1* |
| Handicap Index (HAQ) c | 2.0 | 1.9 | 1.9 | 1,5* | 1.5 | 1.3 | 1.5 | 0,8* |
| CRP (Mg/DL) | 3.9 | 4.3 | 4.6 | 1.8* | 1.0 | 0.9 | 1.0 | 0,4* |
| a 40 mg Humira administreret hver anden uge b Visuel analog skala; 0 = bedste 10 = værst c Handicapindeks for spørgeskemaet for sundhedsvurdering; 0 = Bedste 3 = værste måling * s <0.001 Humira vs. placebo based on mean change from baseline |
Tidsforløbet for ACR 20-respons for undersøgelse RA-III er vist i figur 1.
I undersøgelsen opretholdt RA-III 85% af patienterne med ACR 20-svar i uge 24 responsen efter 52 uger. Tidsforløbet for ACR 20-respons for undersøgelse RA-I og undersøgelse RA-II var ens.
Figur 1: Undersøg RA-III ACR 20-svar over 52 uger
 |
I undersøgelse RA-IV 53% af patienterne behandlet med Humira 40 mg hver anden uge plus plejestandard havde et ACR 20-svar i uge 24 sammenlignet med 35% på placebo plus plejestandard (P <0.001). No unique adverse reactions related to the combination of Humira (adalimumab) og other DMARDs were observed.
I undersøgelse RA-V med MTX NAã¯ve-patienter med nyere begyndelse RA kombinationsbehandlingen med HUMIRA plus MTX førte til større procentdel af patienter, der opnåede ACR-responser end enten MTX-monoterapi eller Humira-monoterapi i uge 52, og svarene blev opretholdt i uge 104 (se tabel 5).
Tabel 5: ACR-respons i undersøgelse RA-V (procent af patienterne)
| Svar | MTX b N = 257 | Humira c N = 274 | Humira/MTX N = 268 |
| ACR20 | |||
| Uge 52 | 63% | 54% | 73% |
| Uge 104 | 56% | 49% | 69% |
| ACR50 | |||
| Uge 52 | 46% | 41% | 62% |
| Uge 104 | 43% | 37% | 59% |
| ACR70 | |||
| Uge 52 | 27% | 26% | 46% |
| Uge 104 | 28% | 28% | 47% |
| Større klinisk respons a | 28% | 25% | 49% |
| a Større klinisk respons defineres som at opnå et ACR70 -svar i en kontinuerlig seks måneders periode b p <0.05 Humira/MTX vs. MTX for ACR 20 p <0.001 Humira/MTX vs. MTX for ACR 50 og 70 og Større klinisk respons c p <0.001 Humira/MTX vs. Humira |
I uge 52 blev alle individuelle komponenter i ACR-responskriterierne for undersøgelse RA-V forbedret i Humira/MTX-gruppen, og forbedringer blev opretholdt til uge 104.
Radiografisk respons
I undersøgelsen blev RA-III strukturel ledskade vurderet radiografisk og udtrykt som ændring i total skarp score (TSS) og dens komponenter erosions score og fælles plads indsnævring (JSN) score ved måned 12 sammenlignet med baseline. Ved baseline var median TSS ca. 55 i placebo og 40 mg hver anden uges grupper. Resultaterne er vist i tabel 6. HUMIRA/MTX -behandlede patienter demonstrerede mindre radiografisk progression end patienter, der fik MTX alene efter 52 uger.
Tabel 6: Radiografiske gennemsnitlige ændringer over 12 måneder i studiet RA-III
| Placebo/MTX | Humira/MTX 40 mg hver anden uge | Placebo/MTX-Humira/MTX (95% Confidence Interval*) | P-værdi ** | |
| Total skarp score | 2.7 | 0.1 | 2.6 (NULL,4 3.8) | <0.001 |
| Erosion score | 1.6 | 0.0 | 1,6 (NULL,9 2.2) | <0.001 |
| JSN -score | 1.0 | 0.1 | 0,9 (NULL,3 1,4) | 0.002 |
| *95% konfidensintervaller for forskellene i ændringsresultater mellem MTX og Humira. ** Baseret på ranganalyse |
I den open-label-udvidelse af undersøgelsen blev RA-III 77% af de originale patienter behandlet med enhver dosis Humira evalueret radiografisk efter 2 år. Patienter opretholdt inhibering af strukturel skade målt ved TSS. Fireogtredive procent havde ingen progression af strukturel skade som defineret ved en ændring i TSS på nul eller mindre. Femogtyve procent (55%) af patienterne, der oprindeligt blev behandlet med 40 mg Humira hver anden uge, er blevet evalueret radiografisk efter 5 år. Patienterne havde fortsat inhibering af strukturelle skader uden 50%, der ikke viser nogen progression af strukturel skade defineret ved en ændring i TSS for nul eller mindre.
I undersøgelsen blev RA-V-strukturelle ledskader vurderet som i undersøgelse RA-III. Større inhibering af radiografisk progression som vurderet ved ændringer i TSS -erosionsscore og JSN blev observeret i Humira/MTX -kombinationsgruppen sammenlignet med enten MTX- eller Humira -monoterapi -gruppen i uge 52 såvel som i uge 104 (se tabel 7).
Tabel 7: Radiografisk gennemsnitlig ændring* I studiet RA-V
| MTX a N = 257 | Humira ab N = 274 | Humira/MTX N = 268 | ||
| 52 uger | Total skarp score | 5.7 (4.2 7.3) | 3.0 (NULL,7 4.3) | 1,3 (NULL,5 2.1) |
| Erosion score | 3.7 (2.7 4.8) | 1,7 (NULL,0 2.4) | 0,8 (NULL,4 1,2) | |
| JSN -score | 2,0 (NULL,2 2.8) | 1,3 (NULL,5 2.1) | 0,5 (NULL,0 1,0) | |
| 104 uger | Total skarp score | 10.4 (7.7 13.2) | 5,5 (NULL,6 7,4) | 1,9 (NULL,9 2.9) |
| Erosion score | 6.4 (4.6 8.2) | 3.0 (2.0 4.0) | 1,0 (NULL,4 1,6) | |
| JSN -score | 4.1 (NULL,7 5.4) | 2.6 (NULL,5 3.7) | 0,9 (NULL,3 1,5) | |
| * Gennemsnit (95% konfidensinterval) a p <0.001 Humira/MTX vs. MTX at 52 og 104 weeks og for Humira/MTX vs. Humira at 104 weeks b p <0.01 for Humira/MTX vs. Humira at 52 uger |
Fysisk funktionsrespons
I undersøgelser viste RA-I gennem IV Humira signifikant større forbedring end placebo i handicapindekset for spørgeskemaet for sundhedsvurdering (HAQ-DI) fra baseline til slutningen af studiet og signifikant større forbedring end placebo i sundhedsudviklingerne som vurderet af Short Form Health Survey (SF 36). Forbedring blev set i både den fysiske komponentoversigt (PCS) og den mentale komponentoversigt (MCS).
I undersøgelsen af RA-III var den gennemsnitlige (95% CI) forbedring i HAQ-DI fra baseline i uge 52 0,60 (NULL,55 0,65) for Humira-patienterne og 0,25 (NULL,17 0,33) for placebo/MTX (P <0.001) patients. Sixty-three percent of Humira-treated patients achieved a 0.5 or greater improvement in HAQ-DI at week 52 in the double-blind portion of the study. Eighty-two percent of these patients maintained that improvement through week 104 og a similar proportion of patients maintained this response through week 260 (5 years) of open-label treatment. Mean improvement in the SF-36 was maintained through the end of measurement at week 156 (3 years).
I studiet viste Ra-V Haq-di og den fysiske komponent i SF-36 større forbedring (P <0.001) for the Humira/MTX combination therapy group versus either the MTX moikkeherapy or the Humira moikkeherapy group at Uge 52 which was maintained through Uge 104.
Juvenil idiopatisk arthritis
Sikkerheden og effektiviteten af Humira blev vurderet i to undersøgelser (undersøgelser Jia-I og Jia-II) hos patienter med aktiv polyartikulær juvenil idiopatisk arthritis (JIA).
Undersøg Jia-i
Sikkerheden og effektiviteten af HUMIRA blev vurderet i et multicenter randomiseret tilbagetrækning dobbeltblind parallel-gruppestudie hos 171 patienter, der var 4 til 17 år med polyartikulær JIA. I undersøgelsen blev patienterne stratificeret i to grupper: MTX-behandlet eller ikke-MTX-behandlet. Alle patienter måtte vise tegn på aktiv moderat eller alvorlig sygdom på trods af tidligere behandling med NSAIDS -smertestillende kortikosteroider eller DMARD'er. Patienter, der modtog forudgående behandling med biologiske DMARD'er, blev udelukket fra undersøgelsen.
Undersøgelsen omfattede fire faser: en åben mærket føring i fase (OL-LI; 16 uger) en dobbeltblind randomiseret tilbagetrækningsfase (DB; 32 uger) en åben-mærket forlængelsesfase (OLE-BSA; op til 136 uger) og en åben mærket fast dosisfase (OLE-FD; 16 uger). I de første tre faser af undersøgelsen blev Humira administreret baseret på kropsoverfladeareal i en dosis på 24 mg/m² op til en maksimal total kropsdosis på 40 mg subkutant (SC) hver anden uge. I ole-FD-fasen blev patienterne behandlet med 20 mg Humira SC hver anden uge, hvis deres vægt var mindre end 30 kg og med 40 mg Humira SC hver anden uge, hvis deres vægt var 30 kg eller mere. Patienter forblev på stabile doser af NSAID'er og eller prednison (≤0,2 mg/kg/dag eller 10 mg/dag maksimalt).
Patienter, der demonstrerede en pædiatrisk ACR 30-respons i slutningen af OL-LI-fasen, blev randomiseret til den dobbelte blinde (DB) fase af undersøgelsen og modtog enten Humira eller placebo hver anden uge i 32 uger eller indtil sygdommen flare. Sygdomsflare blev defineret som en forværring af ≥30% fra baseline i ≥3 af 6 pædiatriske ACR -kernerskriterier ≥2 aktive led og forbedring af> 30% i højst 1 af de 6 kriterier. Efter 32 uger eller på tidspunktet for sygdom blev blussen under DB-fasen behandlet i den åbne labelforlængelsesfase baseret på BSA-regimet (OLBSA), før de konverterede til et fast dosisregime baseret på kropsvægt (OLE-FD-fase).
Undersøg Jia-i Klinisk respons
I slutningen af den 16-ugers OL-LI-fase 94% af patienterne i MTX-stratumet og 74% af patienterne i ikke-MTX-stratumet var pædiatriske ACR 30-respondere. I DB -fasen oplevede signifikant færre patienter, der modtog Humira, sygdomsflare sammenlignet med placebo både uden MTX (43% mod 71%) og med MTX (37% mod 65%). Flere patienter behandlet med Humira fortsatte med at vise pædiatrisk ACR 30/50/70 -svar i uge 48 sammenlignet med patienter behandlet med placebo. Pædiatriske ACR -svar blev opretholdt i op til to år i OLE -fasen hos patienter, der modtog Humira gennem hele undersøgelsen.
Undersøg Jia-iI
Humira was assessed in an open-label multicenter study in 32 patients who were 2 to <4 years of age or 4 years of age og older weighing <15 kg with moderately to severely active polyarticular JIA. Most patients (97%) received at least 24 uger of Humira treatment dosed 24 mg/m² up to a maximum of 20 mg hver anden uge as a single SC injection up to a maximum of 120 weeks duration. During the study most patients used concomitant MTX with fewer reporting use of corticosteroids or NSAIDs. The primary objective of the study was evaluation of safety [see Bivirkninger ].
Psoriasis arthritis
Sikkerheden og effektiviteten af Humira blev vurderet i to randomiserede dobbeltblinde placebo-kontrollerede studier hos 413 patienter med psoriasisartritis (PSA). Efter afslutningen af begge undersøgelser blev 383 patienter indskrevet i en open-label-udvidelsesundersøgelse, hvor 40 mg Humira blev administreret hver anden uge.
Undersøg PSA-I tilmeldte 313 voksne patienter med moderat til alvorligt aktiv PSA (> 3 hævede og> 3 ømme led), der havde en utilstrækkelig respons på NSAID-terapi i en af følgende former: (1) distale interfalangeale (dip) involvering (n = 23); (2) polyartikulær arthritis (fravær af reumatoidknudler og tilstedeværelse af plaque psoriasis) (n = 210); (3) arthritis -lemlødninger (n = 1); (4) asymmetrisk PSA (n = 77); eller (5) as-lignende (n = 2). Patienter på MTX -terapi (158 af 313 patienter) ved tilmelding (stabil dosis på ≤30 mg/uge i> 1 måned) kunne fortsætte MTX i den samme dosis. Doser af Humira 40 mg eller placebo hver anden uge blev administreret i løbet af den 24-ugers dobbeltblinde periode i undersøgelsen.
Sammenlignet med placebo -behandling med Humira resulterede i forbedringer i målingerne af sygdomsaktivitet (se tabel 8 og 9). Blandt patienter med PSA, der modtog Humira, var de kliniske responser tydelige hos nogle patienter på tidspunktet for det første besøg (to uger) og blev opretholdt op til 88 uger i den igangværende open-label-undersøgelse. Lignende responser blev set hos patienter med hver af undertyperne af psoriasis arthritis, skønt få patienter blev tilmeldt arthritis-luetilanerne og ankyloserende spondylitis-lignende undertyper. Svarene var ens hos patienter, der ikke modtog samtidig MTX -terapi ved baseline.
Patienter med psoriasisk involvering af mindst tre procent kropsoverfladeareal (BSA) blev evalueret for psoriasisområde og sværhedsindeks (PASI) svar. Efter 24 uger var andelene af patienter, der opnåede en forbedring på 75% eller 90% i PASI, henholdsvis 59% og 42% i Humira -gruppen (n = 69) sammenlignet med henholdsvis 1% og 0% i placebogruppen (n = 69) (p <0.001). PASI responses were apparent in some patients at the time of the first visit (two weeks). Svars were similar in patients who were or were ikke receiving concomitant MTX therapy at baseline.
Tabel 8: ACR-respons i undersøgelse PSA-I (procent af patienterne)
| Placebo N = 162 | Humira* N = 151 | |
| ACR20 | ||
| Uge 12 | 14% | 58% |
| Uge 24 | 15% | 57% |
| ACR50 | ||
| Uge 12 | 4% | 36% |
| Uge 24 | 6% | 39% |
| ACR70 | ||
| Uge 12 | 1% | 20% |
| Uge 24 | 1% | 23% |
| * s <0.001 for all comparisons between Humira og placebo |
Tabel 9: Komponenter i sygdomsaktivitet i undersøgelse PSA-I
| Parameter: Median | Placebo N = 162 | Humira* N = 151 | ||
| Baseline | 24 uger | Baseline | 24 uger | |
| Antal ømme led a | 23.0 | 17.0 | 20.0 | 5.0 |
| Antal hævede led b | 11.0 | 9.0 | 11.0 | 3.0 |
| Læge Global vurdering c | 53.0 | 49.0 | 55.0 | 16.0 |
| Patientens globale vurdering c | 49.5 | 49.0 | 48.0 | 20.0 |
| Smerte c | 49.0 | 49.0 | 54.0 | 20.0 |
| Handicap Index (HAQ) d | 1.0 | 0.9 | 1.0 | 0.4 |
| CRP (Mg/DL) e | 0.8 | 0.7 | 0.8 | 0.2 |
| * s <0.001 for Humira vs. placebo comparisons based on median changes a Skala 0-78 b Skala 0-76 c Visuel analog skala; 0 = Bedste 100 = værst d Handicapindeks for spørgeskemaet for sundhedsvurdering; 0 = Bedste 3 = værst; Foranstaltninger Patientens evne til at udføre følgende: Kjole/brudgom opstår Eat Walk Right Grip Oprethold hygiejne og oprethold den daglige aktivitet. e Normalt interval: 0-0.287 mg/dl |
Lignende resultater blev set i en yderligere 12-ugers undersøgelse hos 100 patienter med moderat til svær psoriasisartrit, der havde suboptimal respons på DMARD-terapi som manifesteret ved ≥3 ømme led og ≥3 hævede led ved tilmelding.
Radiografisk respons
Radiografiske ændringer blev vurderet i PSA -undersøgelserne. Radiografer af hænder håndled og fødder blev opnået ved baseline og uge 24 i den dobbeltblinde periode, hvor patienterne var på Humira eller placebo og i uge 48, da alle patienter var på open-label Humira. En modificeret total skarp score (MTSS), der omfattede distale interphalangeale led (dvs. ikke identisk med de TSS, der blev anvendt til reumatoid arthritis) blev anvendt af læsere, der er blindet til behandlingsgruppen for at vurdere røntgenbillederne.
Humira-treated patients demonstrated greater inhibition of radiographic progression comparød to placebo-treated patients og this effect was maintained at 48 uger (see Table 10).
Tabel 10: Ændring i modificeret total skarp score i psoriasisartritis
| Placebo N = 141 | Humira N = 133 | ||
| Uge 24 | Uge 24 | Uge 48 | |
| Baseline mean | 22.1 | 23.4 | 23.4 |
| Gennemsnitlig ændring ± SD | 0,9 ± 3,1 | -0,1 ± 1,7 | -0,2 ± 4,9* |
| * <0.001 for the difference between Humira Uge 48 og Placebo Uge 24 (primary analysis) |
Fysisk funktionsrespons
I undersøgelsen blev PSA-I fysisk funktion og handicap vurderet ved hjælp af HAQ Disability Index (HAQ-DI) og SF-36 Health Survey. Patienter, der blev behandlet med 40 mg Humira hver anden uge, viste større forbedring fra basislinjen i HAQ-DI-score (gennemsnitlige fald på henholdsvis 47% og 49% i uger 12 og 24) i sammenligning med placebo (gennemsnitlige fald på henholdsvis 1% og 3% i uge 12 og 24). I uger 12 og 24 patienter, der blev behandlet med HUMIRA, viste større forbedring fra baseline i SF-36 fysisk komponentoversigts score sammenlignet med patienter, der blev behandlet med placebo og ingen forværring i SF-36 Mental Component Summary Summary Resultat. Forbedring i fysisk funktion baseret på HAQ-DI blev opretholdt i op til 84 uger gennem den åbne del af undersøgelsen.
Ankyloserende spondylitis
Sikkerheden og effektiviteten af Humira 40 mg hver anden uge blev vurderet hos 315 voksne patienter i en randomiseret 24 ugers dobbeltblind placebo-kontrolleret undersøgelse hos patienter med aktiv ankyloserende spondylitis (AS), der havde en utilstrækkelig respons på glukokortikoider NSAIDS-analgetiske methotrexat eller sulfasalazine. Aktiv som blev defineret som patienter, der opfyldte mindst to af de følgende tre kriterier: (1) et bad som sygdomsaktivitetsindeks (BASDAI) score ≥4 cm (2) en visuel analog score (VAS) for total rygsmerter ≥ 40 mm og (3) morgenstivhed ≥ 1 time. Den blindede periode blev efterfulgt af en open-label-periode, hvor patienterne modtog Humira 40 mg hver anden uge subkutant i op til yderligere 28 uger.
Forbedring af målinger af sygdomsaktivitet blev først observeret i uge 2 og opretholdt gennem 24 uger som vist i figur 2 og tabel 11.
Svars of patients with total spinal ankylosis (n=11) were similar to those without total ankylosis.
Figur 2: ASAS 20 Respons ved besøgsundersøgelse som-I
 |
Efter 12 uger blev ASAS 20/50/70 svar opnået med henholdsvis 58% 38% og 23% af patienter, der fik HUMIRA sammenlignet med henholdsvis 21% 10% og 5% af patienter, der modtog placebo (P <0.001). Similar responses were seen at Uge 24 og were sustained in patients receiving open-label Humira for up to 52 uger.
En større andel af patienter, der blev behandlet med Humira (22%), opnåede et lavt niveau af sygdomsaktivitet efter 24 uger (defineret som en værdi <20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) comparød to patients treated with placebo (6%).
Tabel 11: Komponenter i ankyloserende spondylitis sygdomsaktivitet
| Placebo N = 107 | Humira N = 208 | |||
| Baseline mean | Uge 24 mean | Baseline mean | Uge 24 mean | |
| Vinger 20 kriterier svar* | ||||
| Patientens globale vurdering af sygdomsaktivitet a * | 65 | 60 | 63 | 38 |
| Total rygsmerter* | 67 | 58 | 65 | 37 |
| Betændelse b * | 6.7 | 5.6 | 6.7 | 3.6 |
| Basfi c * | 56 | 51 | 52 | 34 |
| Basdai d score* | 6.3 | 5.5 | 6.3 | 3.7 |
| Basmi e score* | 4.2 | 4.1 | 3.8 | 3.3 |
| Tragus til væg (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Lumbar Flexion (CM) | 4.1 | 4.0 | 4.2 | 4.4 |
| Cervikal rotation (grader) | 42.2 | 42.1 | 48.4 | 51.6 |
| Lumbin Side Flexion (CM) | 8.9 | 9.0 | 9.7 | 11.7 |
| Intermalleolær afstand (CM) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRP f * | 2.2 | 2.0 | 1.8 | 0.6 |
| a Procent af personer med mindst en forbedring på 20% og 10-enheder målt på en visuel Analog skala (VAS) med 0 = ingen og 100 = svær b Gennemsnit af spørgsmål 5 og 6 i Basdai (defineret i â € ˜Dâ € ™) c Bath Ankylosing spondylitis funktionelt indeks d Bath Ankylosing Spondylitis Disease Activity Index e Bath Ankylosing Spondylitis Metrology Index f C-reaktivt protein (Mg/DL) * statistisk signifikant for sammenligninger mellem Humira og placebo i uge 24 |
En anden randomiseret multicenter dobbeltblind placebokontrolleret undersøgelse af 82 patienter med ankyloserende spondylitis viste lignende resultater.
Patienter, der blev behandlet med Humira, opnåede forbedringer fra baseline i den ankyloserende spondylitis livskvalitet for livskvalitet (ASQOL) score (-3,6 vs. -1,1) og i den korte form sundhedsundersøgelse (SF-36) fysisk komponentoversigt (PCS) score (NULL,4 vs. 1,9) sammenlignet med placebo-behandlingspatienter i uge 24.
Voksen Crohns sygdom
Sikkerheden og effektiviteten af flere doser af Humira blev vurderet hos voksne patienter med moderat til alvorligt aktiv Crohns sygdom-CD (Crohns sygdomsaktivitetsindeks (CDAI) ≥ 220 og ≤ 450) i randomiserede dobbeltblinde placebokontrollerede studier. Samtidig stabile doser af aminosalicylater kortikosteroider og/eller immunmodulerende midler var tilladt, og 79% af patienterne fortsatte med at modtage mindst en af disse medicin.
Induktion af klinisk remission (defineret som CDAi <150) was evaluated in two studies. In Study CD-I 299 TNF-blocker naïve patients were rogomized to one of four treatment groups: the placebo group received placebo at Weeks 0 og 2 the 160/80 group received 160 mg Humira at Week 0 og 80 mg at Week 2 the 80/40 group received 80 mg at Week 0 og 40 mg at Week 2 og the 40/20 group received 40 mg at Week 0 og 20 mg at Week 2. Clinical results were assessed at Uge 4.
I den anden induktionsundersøgelsesundersøgelse CD-II 325 patienter, der havde mistet responsen på eller var intolerante over for tidligere infliximab-terapi, blev randomiseret til at modtage enten 160 mg Humira i uge 0 og 80 mg i uge 2 eller placebo i uge 0 og 2. kliniske resultater blev vurderet i uge 4.
Vedligeholdelse af klinisk remission blev evalueret i undersøgelse CD-III. I denne undersøgelse modtog 854 patienter med aktiv sygdom åben mærket Humira 80 mg i uge 0 og 40 mg i uge 2.. Patienter blev derefter randomiseret i uge 4 til 40 mg Humira hver anden uge 40 mg Humira hver uge eller placebo. Den samlede undersøgelsesvarighed var 56 uger. Patienter i klinisk respons (fald i CDAI ≥70) i uge 4 blev lagdelt og analyseret separat fra dem, der ikke var i klinisk respons i uge 4.
Induktion af klinisk remission
En større procentdel af de patienter, der blev behandlet med 160/80 mg HUMIRA, opnåede induktion af klinisk remission versus placebo i uge 4 uanset om patienterne var TNF-blokkering Naã¯ve (CD-I) eller havde mistet respons på eller var intolerante over for infliximab (CD-II) (se tabel 12).
Tabel 12: Induktion af klinisk remission i studier CD-I og CD-II (procent af patienterne)
| CD-I | CD-II | |||
| Placebo N = 74 | Humira 160/80 mg N = 76 | Placebo N = 166 | Humira 160/80 mg N = 159 | |
| Uge 4 | ||||
| Klinisk remission | 12% | 36%* | 7% | 21%* |
| Klinisk respons | 34% | 58%** | 34% | 52%** |
| Klinisk remission is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. * s <0.001 for Humira vs. placebo pairwise comparison of proportions ** s <0.01 for Humira vs. placebo pairwise comparison of proportions |
Vedligeholdelse af klinisk remission
I undersøgelse var CD-III i uge 4 58% (499/854) af patienter i klinisk respons og blev vurderet i den primære analyse. I uger 26 og 56 opnåede større andele af patienter, der var i klinisk respons i uge 4, klinisk remission i Humira 40 mg hver anden uges vedligeholdelsesgruppe sammenlignet med patienter i placebo -vedligeholdelsesgruppen (se tabel 13). Gruppen, der modtog Humira -terapi hver uge, demonstrerede ikke signifikant højere remissionsrater sammenlignet med gruppen, der modtog Humira hver anden uge.
Tabel 13: Vedligeholdelse af klinisk remission i CD-III (procent af patienterne)
| Placebo N = 170 | 40 mg Humira hver anden uge N = 172 | |
| Uge 26 | ||
| Klinisk remission | 17% | 40%* |
| Klinisk respons | 28% | 54%* |
| Uge 56 | ||
| Klinisk remission | 12% | 36%* |
| Klinisk respons | 18% | 43%* |
| Klinisk remission is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. *s <0.001 for Humira vs. placebo pairwise comparisons of proportions |
Af dem som svar i uge 4, der opnåede remission under undersøgelsespatienterne i Humira hver anden uges gruppe opretholdt remission i længere tid end patienter i placebo -vedligeholdelsesgruppen. Blandt patienter, der ikke var som svar ved uge 12 -terapi, fortsatte over 12 uger, resulterede ikke i signifikant flere svar.
Pædiatrisk Crohns sygdom
En randomiseret dobbeltblind 52-ugers klinisk undersøgelse af 2 dosiskoncentrationer af HUMIRA (undersøgelse PCD-I) blev udført i 192 pædiatriske patienter (6 til 17 år) med moderat til alvorligt aktiv Crohns sygdom (defineret som pædiatrisk Crohns sygdomsaktivitetsindeks (PCDAI) score> 30). Tilmeldte patienter havde i løbet af den foregående toårsperiode en utilstrækkelig respons på kortikosteroider eller en immunmodulator (dvs. azathioprin 6-mercaptopurin eller methotrexat). Patienter, der tidligere havde modtaget en TNF -blokkering, fik lov til at tilmelde sig, hvis de tidligere havde haft tab af respons eller intolerance over for den TNF -blokering.
Patienter modtog open-label-induktionsterapi i en dosis baseret på deres kropsvægt (≥40 kg og <40 kg). Patients weighing ≥40 kg received 160 mg (at Week 0) og 80 mg (at Week 2). Patients weighing <40 kg received 80 mg (at Week 0) og 40 mg (at Week 2). At Uge 4 patients within each body weight category (≥40 kg og <40 kg) were rogomized 1:1 to one of two maintenance dose regimens (high dose og low dose). The high dose was 40 mg hver anden uge for patients weighing ≥40 kg og 20 mg hver anden uge for patients weighing <40 kg. The low dose was 20 mg hver anden uge for patients weighing ≥40 kg og 10 mg hver anden uge for patients weighing <40 kg.
Samtidig stabile doser af kortikosteroider (prednisondosering ≤40 mg/dag eller tilsvarende) og immunmodulatorer (azathioprin 6-mercaptopurin eller methotrexat) var tilladt i hele undersøgelsen.
I uge 12 patienter, der oplevede en sygdomsflare (stigning i PCDAI på ≥ 15 fra uge 4 og absolut PCDAI> 30) eller som ikke var responderende (opnåede ikke et fald i PCDAI på ≥ 15 fra baseline i 2 på hinanden følgende besøg mindst 2 ugers mellemrum) fik dosis-escalat (i.e. switch fra blindt hver anden uge dosering til blind hver uge dosering); Patienter, der dosisbelagte, blev betragtet som behandlingsfejl.
Ved baseline modtog 38% af patienterne kortikosteroider, og 62% af patienterne modtog en immunmodulator. 42 procent (44%) af patienterne havde tidligere mistet responsen eller var intolerante over for en TNF-blokering. Den median baseline PCDAI -score var 40.
Af de 192 patienter afsluttede i alt 188 patienter den 4 ugers induktionsperiode 152 patienter afsluttede 26 ugers behandling og 124 patienter afsluttede 52 ugers behandling. Femoghalvfjerds procent (51%) (48/95) af patienter i dosisdosisdosis-dosis-dosis og 38% (35/93) af patienter i dosisdosis-dosis-dosis-dosis.
I uge 4 28% (52/188) af patienter var i klinisk remission (defineret som PCDAI ≤ 10).
Proportionerne af patienter i klinisk remission (defineret som PCDAI ≤ 10) og klinisk respons (defineret som reduktion i PCDAI på mindst 15 point fra baseline) blev vurderet i uger 26 og 52.
På både uger 26 og 52 var andelen af patienter i klinisk remission og klinisk respons numerisk højere i gruppen med høj dosis sammenlignet med gruppen med lav dosis (tabel 14). Det anbefalede vedligeholdelsesregime er 20 mg hver anden uge for patienter, der vejer <40 kg og 40 mg hver anden uge for patients weighing ≥ 40 kg. Every week dosing is ikke the recommended maintenance dosing regimen [see Dosering og administration ].
Tabel 14: Klinisk remission og klinisk respons i undersøgelse PCD-I
| Lav vedligeholdelsesdosis † (20 eller 10 mg hver anden uge) N = 95 | Høj vedligeholdelsesdosis N = 93 | |
| Uge 26 | ||
| Klinisk remission ‡ | 28% | 39% |
| Klinisk respons§ | 48% | 59% |
| Uge 52 | ||
| Klinisk remission ‡ | 23% | 33% |
| Klinisk respons§ | 28% | 42% |
| † Den lave vedligeholdelsesdosis var 20 mg hver anden uge for patienter, der vejer ≥ 40 kg og 10 mg hver anden uge for patienter, der ligger <40 kg. <40 kg. ‡ Klinisk remission defineret som PCDAI ≤ 10. §Clinical respons defineret som reduktion i PCDAI på mindst 15 point fra baseline. |
Voksen ulcerøs colitis
The safety and efficacy of HUMIRA were assessed in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12 on a 12 point scale with an endoscopy subscore of 2 to 3 on a scale of 0 to 3) despite concurrent or prior treatment with immunosuppressants such as corticosteroids azathioprine or 6-MP in two randomized double-blind placebo-controlled clinical studies (Studies UC-I og UC-II). Begge undersøgelser tilmeldte TNF-blokkering NAã¯ve-patienter, men undersøgte UC-II tilladte også indtræden af patienter, der mistede respons på eller var intolerante over for TNFBLOCKERS. Fyrre procent (40%) af patienter, der var indskrevet i undersøgelse UC-II, havde tidligere brugt en anden TNF-blokkering.
Samtidig stabile doser af aminosalicylater og immunsuppressiva var tilladt. I undersøgelser modtog UC-I- og II-patienter aminosalicylater (69%) kortikosteroider (59%) og/eller azathioprin eller 6-MP (37%) ved baseline. I begge undersøgelser modtog 92% af patienterne mindst en af disse medicin.
Induktion af klinisk remission (defineret som mayo -score ≤ 2 uden nogen individuelle undercores> 1) i uge 8 blev evalueret i begge undersøgelser. Klinisk remission i uge 52 og vedvarende klinisk remission (defineret som klinisk remission ved både uger 8 og 52) blev evalueret i undersøgelse UC-II.
I undersøgelse blev UC-I 390 TNF-blokker NAã¯ve-patienter randomiseret til en af tre behandlingsgrupper til den primære effektivitetsanalyse. Placebogruppen modtog placebo i uger 0 2 4 og 6. 160/80 -gruppen modtog 160 mg Humira i uge 0 og 80 mg i uge 2, og 80/40 -gruppen modtog 80 mg Humira i uge 0 og 40 mg i uge 2. efter uge 2 patienter i begge Humira -behandlingsgrupper modtog 40 mg hver anden uge.
I undersøgelse blev UC-II 518 patienter randomiseret til at modtage enten Humira 160 mg i uge 0 80 mg i uge 2 og 40 mg hver anden uge fra uge 4 til uge 50 eller placebo, der starter ved uge 0 og hver anden uge til uge 50. Corticosteroid Taper var tilladt fra uge 8.
I begge undersøgelser UC-I og UC-II er en større procentdel af de patienter, der blev behandlet med 160/80 mg Humira sammenlignet med patienter behandlet med placebo, induktion af klinisk remission. I undersøgelse UC-II en større procentdel af de patienter, der blev behandlet med 160/80 mg HUMIRA sammenlignet med patienter behandlet med placebo, opnåede vedvarende klinisk remission (klinisk remission i både uger 8 og 52) (tabel 15).
Tabel 15: Induktion af klinisk remission i undersøgelser UC-I og UC-II og vedvarende klinisk remission i undersøgelse UC-II (procent af patienterne)
| Undersøg UC-I | Undersøg UC-II | |||||
| Placebo N = 130 | Humira 160/80 mg N = 130 | Behandlingsforskel (95% CI) | Placebo N = 246 | Humira 160/80 mg N = 248 | Behandlingsforskel (95% CI) | |
| Induktion af klinisk remission (klinisk remission i uge 8) | 9,2% | 18,5% | 9,3%* (NULL,9%17,6%) | 9,3% | 16,5% | 7,2%* (NULL,2%12,9%) |
| Vedvarende klinisk remission (klinisk remission ved både uger 8 og 52) | N/a | N/a | N/a | 4,1% | 8,5% | 4,4%* (NULL,1%8,6%) |
| Klinisk remission defineres som mayo -score ≤ 2 uden nogen individuelle undercores> 1. CI = konfidensinterval * s <0.05 for Humira vs. placebo pairwise comparison of proportions |
I undersøgelse UC-I var der ingen statistisk signifikant forskel i klinisk remission observeret mellem Humira 80/40 mg-gruppen og placebogruppen i uge 8.
I undersøgelse UC-II 17,3% (43/248) i Humira-gruppen var i klinisk remission i uge 52 sammenlignet med 8,5% (21/246) i placebogruppen (behandlingsforskel: 8,8%; 95% konfidensinterval (CI): [2,8% 14,5%]; <0.05).
I undergruppen af patienter i undersøgelsen UC-II med tidligere TNF-blokkering syntes brugen af behandlingsforskellen til induktion af klinisk remission at være lavere end den, der blev set i undersøgelsespopulationen, og behandlingsforskellene for vedvarende klinisk remission og klinisk remission i uge 52 syntes at svare til dem, der blev set i hele undersøgelsespopulationen. Undergruppen af patienter med tidligere TNF-blokkeringsanvendelse opnåede induktion af klinisk remission ved 9% (9/98) i Humira-gruppen mod 7% (7/101) i placebogruppen og vedvarende klinisk remission ved 5% (5/98) i Humira-gruppen mod 1% (1/101) i placebogruppen. I undergruppen af patienter med tidligere TNF-blokkering var brug af 10% (10/98) i klinisk remission i uge 52 i Humira-gruppen mod 3% (3/101) i placebogruppen.
Pædiatrisk ulcerøs colitis
Sikkerheden og effektiviteten af Humira blev vurderet i et multicenter randomiseret dobbeltblindt forsøg (undersøgelse PUC-I NCT02065557) hos 93 pædiatriske patienter 5 år til 17 år med moderat til alvorligt aktiv ulcerøs colitis (Mayo score 6 til 12 med endoskopi-del af 2 til 3 point bekræftet ved centralt læsning af endoskopi), hvem med kortikosteroider og/eller en immunmodulator (dvs. azathioprin 6mercaptopurin eller methotrexat). Femten ud af 93 patienter (16%) i undersøgelsen havde tidligere erfaring med en TNF -blokering. Patienter, der modtog kortikosteroider ved tilmelding, fik lov til at tilspidses deres kortikosteroidbehandling efter uge 4.
Syvogtyve patienter blev oprindeligt randomiseret 3: 2 for at modtage dobbeltblind behandling med en af to doser af Humira. Patienter i begge doseringsgrupper modtog 2,4 mg/kg (maksimalt 160 mg) ved uge 0 1,2 mg/kg (maksimalt 80 mg) i uge 2 og 0,6 mg/kg (maksimalt 40 mg) i uge 4 og 6. Den højere doseringsgruppe modtog også en yderligere dosering på 2,4 mg/kg (maksimum på 160 mg) i uge 1. Efter en ændring af undersøgelsen til undersøgelsen 16 blev tilmeldt og tilmeldt, og det blev tilmeldt åbent, og det blev tilmeldt. Behandling med Humira ved den højere dosering.
I uge 8 62 patienter, der demonstrerede klinisk respons pr. Delvis mayo-score (PMS; en undergruppe af mayo-score uden endoskopisk komponent og defineret som et fald i PMS ≥ 2 point og ≥ 30% fra baseline) blev randomiseret lige for at modtage dobbeltblind behandling med humira 0,6 mg/kg (maksimal 40 mg) hver anden uge (lavere doseringsgruppe) eller 0,6 mg/kg (maksimum på 40 mg) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge) hver uge skal dualen) hver uge) eller lavere dosering) eller 0,6 mg/0,6 mg/maksimum på 40 maks. (Højere doseringsgruppe). Før en ændring af undersøgelsesdesignet blev 12 yderligere patienter, der demonstrerede klinisk respons pr. PMS, randomiseret til at modtage placebo.
Der er ingen forventede klinisk relevante forskelle i effektivitet mellem den studerede højere dosering, der administreres under den 52-ugers PUC-I-forsøg og den anbefalede dosering af Humira [se Dosering og administration Klinisk farmakologi ].
Patienter, der opfyldte kriterierne for sygdom, der blev blusset på eller efter uge 12, blev randomiseret til at modtage en reinduktionsdosis på 2,4 mg/kg (maksimalt 160 mg) eller en dosis på 0,6 mg/kg (maksimalt 40 mg) og fortsatte derefter den dosis, de blev randomiseret i uge 8.
Undersøgelsens co-primære endepunkter var klinisk remission pr. PMS (defineret som PMS ≤ 2 og ingen individuel underkore> 1) i uge 8 og klinisk remission pr. Mayo-score (defineret som Mayo-score ≤ 2 og ingen individuel underkore> 1) ved uge 52 hos patienter, der opnåede klinisk respons pr. PMS i uge 8. Secondary Endpoints inkluderede Mayo-score (defineret som en nedgang i majo-scoret af Mayo på Mayo Olt Olt. ≥ 30% fra baseline) i uge 52 i uge kl. 20 respondere endoskopisk forbedring (defineret som en mayo endoskopi underkore ≤ 1) i uge 52 i uge 8 pms respondenter og mayo score remission i uge 52 i uge kl. 8 pm.
Uge 8 resultater
I uge kl. 20 blev der opnået remission med 60% [28/47; 95% konfidensinterval (CI): (44% 74%)] af patienter i den højere doseringsgruppe (ikke inklusive de 16 patienter, der modtager åben mærket højere dosering) og 43% [13/30; 95% CI: (25% 63%)] af patienter i den nedre doseringsgruppe. Resultater fra den højere doseringsgruppe er repræsentative for de forventede resultater med den anbefalede dosering [se Dosering og administration Klinisk farmakologi ].
Uge 52 Results
I uge blev 52 slutpunkter vurderet hos befolkningen af patienter, der modtog dobbeltblind placebo Humira 0,6 mg/kg (maksimalt 40 mg) hver anden uge eller Humira 0,6 mg/kg (maksimal 40 mg) hver uge mellem uge 8 og uge 52 (tabel 16).
Tabel 16: Klinisk respons på klinisk remission og endoskopisk forbedring i uge 52 hos pædiatriske patienter med ulcerøs colitis (undersøgelse PUC-1)
| Placebo a N/N (%) 95% CI | Humira Maximum of 40 mg (0.6 mg/kg) every other week b N/N (%) 95% CI | Humira Maximum of 40 mg (0.6 mg/kg) every week c N/N (%) 95% CI | |
| Klinisk remission in Week 8 PMS responders | 4/12 (33%) (10%65%) | 9/31 (29%) (14%48%) | 14/31 (45%) (27%64%) |
| Klinisk respons in Week 8 PMS responders | 4/12 (33%) (10%65%) | 19/31 (61%) (42%78%) | 21/31 (68%) (49%83%) |
| Endoskopisk forbedring i ugen kl. 20.00 respondere | 4/12 (33%) (10%65%) | 12/31 (39%) (22%58%) | 16/31 (52%) (33%70%) |
| Klinisk remission in Week 8 PMS remitters | 3/8 (38%) (9%76%) | 9/21 (43%) (22%66%) | 10/22 (45%) (24%68%) |
| CI = konfidensinterval a Tolv patienter, der demonstrerede klinisk respons pr. PMS i uge 8, blev randomiseret til at modtage placebo. Der er begrænsninger for tolkbarheden af placebo -data på grund af den lille prøvestørrelse. b Den anden uges dosering, Dosering og administration ]. c Der er ingen forventede klinisk relevante forskelle i effektivitet mellem den undersøgte højere dosering, der blev administreret under det 52-ugers PUC-I-forsøg og den anbefalede dosering af Humira. Bemærk: Patienter med manglende værdier i uge 52 eller som blev randomiseret til at modtage reinduktion eller vedligeholdelsesbehandling på grund af sygdomsflare blev betragtet som ikke-responderende for uge 52 slutpunkter. |
Plaque psoriasis
Sikkerheden og effektiviteten af HUMIRA blev vurderet i randomiserede dobbeltblinde placebokontrollerede studier hos 1696 voksne personer med moderat til svær kronisk plaque psoriasis (PS), der var kandidater til systemisk terapi eller fototerapi.
Undersøgelse PS-I evaluerede 1212 forsøgspersoner med kronisk PS med ≥10% kropsoverfladeareal (BSA) involvering af lægenes globale vurdering (PGA) af mindst moderat sygdomsgrad og psoriasis-område og sværhedsindeks (PASI) ≥12 inden for tre behandlingsperioder. I periode modtog et forsøgspersoner placebo eller humira i en indledende dosis på 80 mg i uge 0 efterfulgt af en dosis på 40 mg hver anden uge, der starter ved uge 1. efter 16 ugers terapipersoner, der opnåede mindst en PASI 75-respons ved uge 16 defineret som en PASI-score forbedring af mindst 75% i forhold til baseline indledte periode B og modtog åben mærket 40 mg Humira hver anden uge.
Efter 17 uger med åben etiketterapipersoner, der opretholdt mindst en PASI 75-respons i uge 33 og oprindeligt blev randomiseret til aktiv terapi i periode A blev genrandomiseret i periode C for at modtage 40 mg Humira hver anden uge eller placebo i yderligere 19 uger. På tværs af alle behandlingsgrupper var den gennemsnitlige baseline PASI -score 19 og baseline -lægens globale vurderings score varierede fra moderat (53%) til alvorlig (41%) til meget alvorlig (6%).
Undersøgelse PS-II evaluerede 99 forsøgspersoner randomiseret til HUMIRA og 48 forsøgspersoner randomiseret til placebo med kronisk plaque psoriasis med ≥10% BSA-involvering og PASI ≥12. Personer modtog placebo eller en indledende dosis på 80 mg Humira i uge 0 efterfulgt af 40 mg hver anden uge fra uge 1 i 16 uger. På tværs af alle behandlingsgrupper var den gennemsnitlige baseline PASI -score 21 og baseline -PGA -score varierede fra moderat (41%) til svær (51%) til meget alvorlig (8%).
Undersøgelser PS-I og II evaluerede andelen af forsøgspersoner, der opnåede klar eller minimal sygdom på 6-punkts PGA-skala og andelen af forsøgspersoner, der opnåede en reduktion i PASI-score på mindst 75% (PASI 75) fra baseline i uge 16 (se tabel 17 og 18).
Derudover vurderede PS-I andelen af forsøgspersoner, der opretholdt en PGA af klar eller minimal sygdom eller en PASI 75-respons efter uge 33 og før uge 52.
Tabel 17: Effektivitetsresultater ved 16 uger i undersøgelse PS-I Antal emner (%)
| Humira 40 mg hver anden uge N = 814 | Placebo N = 398 | |
| PGA: Klar eller minimal * | 506 (62%) | 17 (4%) |
| Efter 75 | 578 (71%) | 26 (7%) |
| * Rydde = ingen plaquehøjde ingen skala plus eller minus hyperpigmentering eller diffus lyserød eller rød farve minimal = mulig, men vanskelig at konstatere, om der er en lille højde af plak over normal hud plus eller minus overfladørhed med noget hvid farve plus eller minus op til rød farve |
Tabel 18: Effektivitetsresultater ved 16 uger i undersøgelse PS-II Antal emner (%)
| Humira 40 mg hver anden uge N = 99 | Placebo N = 48 | |
| PGA: Klar eller minimal * | 70 (71%) | 5 (10%) |
| Efter 75 | 77 (78%) | 9 (19%) |
| * Ryd = ingen plaque -højde ingen skala plus eller minus hyperpigmentering eller diffus lyserød eller rød farve Minimal = muligt, men vanskeligt at undersøge, om der er en lille højde af plak over normal hud plus eller minus overfladørhed med lidt hvid farve plus eller minus op til rød farve |
Derudover blev PS-I-forsøgspersoner på Humira, der opretholdt en PASI 75, rerandomiseret til Humira (n = 250) eller placebo (n = 240) ved uge 33. Efter 52 ugers behandling med Humira-emner på Humira opretholdt effektiviteten sammenlignet med emner, der blev genrandiseret til placebo baseret på vedligeholdelse af PGA af klar eller minimal sygdom (68% vs. 28) or. 75 (79% mod 43%).
I alt 347 stabile respondere deltog i en evaluering af tilbagetrækning og tilbagebetaling i en åbenliv udvidelsesundersøgelse. Median tid til tilbagefald (tilbagegang til PGA -moderat eller værre) var cirka 5 måneder. I tilbagetrækningsperioden oplevede intet individ omdannelse til hverken pustulær eller erythrodermisk psoriasis. I alt 178 forsøgspersoner, der tilbagefaldt geninitieret behandling med 80 mg Humira, derefter 40 mg hver anden uge, der begyndte i uge 1. i uge 16 69% (123/178) af forsøgspersoner, havde et svar på PGA-klar eller minimal.
En randomiseret dobbeltblind undersøgelse (undersøgelse PS-III) sammenlignede effektiviteten og sikkerheden af Humira versus placebo hos 217 voksne individer. Subjects in the study had to have chronic plaque psoriasis of at least moderate severity on the PGA scale fingernail involvement of at least moderate severity on a 5-point Physician’s Global Assessment of Fingernail Psoriasis (PGA-F) scale a Modified Nail Psoriasis Severity Index (mNAPSI) score for the target-fingernail of ≥ 8 and either a BSA involvement of at least 10% eller en BSA-involvering af mindst 5% med en total MNAPSI-score for alle negle på ≥ 20. Emner modtog en initial dosis på 80 mg Humira efterfulgt af 40 mg hver anden uge (startende en uge efter den indledende dosis) eller placebo i 26 uger efterfulgt af open-label Humira-behandling i yderligere 26 uger. Denne undersøgelse evaluerede andelen af personer, der opnåede klar eller minimal vurdering med mindst en 2-grade forbedring på PGA-F-skalaen og andelen af forsøgspersoner, der opnåede mindst en 75% forbedring fra baseline i MNAPSI-score (MNAPSI 75) i uge 26.
I uge 26 opnåede en højere andel af emner i Humira-gruppen end i placebogruppen PGA-F-slutpunktet. Endvidere opnåede en højere andel af emner i Humira -gruppen end i placebogruppen Mnapsi 75 i uge 26 (se tabel 19).
Tabel 19: Effektivitetsresultater efter 26 uger
| Slutpunkt | Humira 40 mg hver anden uge* N = 109 | Placebo N = 108 |
| PGA-F: ≥2-klasse forbedring og klar eller minimal | 49% | 7% |
| Mnaps 75 | 47% | 3% |
| *Personer modtog 80 mg Humira i uge 0 efterfulgt af 40 mg hver anden uge fra uge 1. |
Neglesmerter blev også evalueret, og forbedring af neglesmerter blev observeret i undersøgelse PS-III.
Hidradenitis suppurativ
To randomiserede dobbeltblinde placebokontrollerede undersøgelser (undersøgelser HS-I og II) evaluerede sikkerheden og effektiviteten af Humira i i alt 633 voksne individer med moderat til svær hidradenitis suppurativa (HS) med Hurley fase II eller III sygdom og med mindst 3 abscesser eller inflammatoriske nodules. I begge undersøgelser modtog forsøgspersoner placebo eller Humira i en indledende dosis på 160 mg i uge 0 80 mg i uge 2 og 40 mg hver uge fra uge 4 og fortsatte gennem uge 11. Emner brugte topisk antiseptisk vask dagligt. Samtidig oral antibiotisk anvendelse blev tilladt i undersøgelse HS-II.
Begge undersøgelser evaluerede hidradenitis suppurativa klinisk respons (HISCR) i uge 12. Hiscr blev defineret som mindst en 50% reduktion i total abscess og inflammatorisk knudeantal uden stigning i abscessantal og ingen stigning i dræning af fistelantal i forhold til baseline (se tabel 18). Reduktion i HS-relateret hudsmerter blev vurderet ved anvendelse af en numerisk vurderingsskala hos patienter, der gik ind i undersøgelsen med en indledende baseline-score på 3 eller mere på en 11-punkts skala.
I begge undersøgelser opnåede en højere andel af Humira- end placebo-behandlede forsøgspersoner Hiscr (se tabel 20).
Tabel 20: Effektivitetsresultater efter 12 uger hos personer med moderat til svær hidradenitis suppurativa
| HS Study i | HS Study iI* | |||
| Placebo | Humira 40 mg Weekly | Placebo | Humira 40 mg Weekly | |
| Hidradenitis suppurativ Klinisk respons (HiSCR) | N = 154 40 (26%) | N = 153 64 (42%) | N = 163 45 (28%) | N = 163 96 (59%) |
| *19,3% af forsøgspersoner i undersøgelse HS-II fortsatte baseline oral antibiotikabehandling under undersøgelsen. |
I begge undersøgelser fra uge 12 til uge 35 (periode B) blev forsøgspersoner, der havde modtaget Humira, genrandomiseret til 1 ud af 3 behandlingsgrupper (Humira 40 mg hver uge Humira 40 mg hver anden uge eller placebo). Personer, der var blevet randomiseret til placebo, fik tildelt Humira 40 mg hver uge (undersøgelse HS-I) eller placebo (undersøgelse HS-II).
I periode B -opblussen af HS defineret som ≥25% stigning fra baseline i abscesser og inflammatoriske noduletællinger og med mindst 2 yderligere læsioner blev dokumenteret i 22 (22%) af de 100 forsøgspersoner, der blev trukket tilbage fra Humira -behandling efter det primære effektivitetstidspunkt i to undersøgelser.
Voksen uveitis
Sikkerheden og effektiviteten af HUMIRA blev vurderet hos voksne patienter med ikke-infektiøse mellemliggende posterior og panuveitis eksklusive patienter med isoleret anterior uveitis i to randomiserede dobbeltmaskede placebokontrollerede undersøgelser (UV I og II). Patienter modtog placebo eller Humira i en indledende dosis på 80 mg efterfulgt af 40 mg hver anden uge, der startede en uge efter den indledende dosis. Det primære effektendepunkt i begge undersøgelser var ”tid til behandlingssvigt”.
Behandlingssvigt var et multikomponentresultat defineret som udviklingen af nye inflammatoriske chorioretinale og/eller inflammatoriske nethindevaskulære læsioner en stigning i anterior kammer (AC) cellekvalitet eller glasagtig dis (VH) eller et fald i bedst korrigeret synsstyrke (BCVA).
Undersøgelse UV I evaluerede 217 patienter med aktiv uveitis, mens de blev behandlet med kortikosteroider (oral prednison i en dosis på 10 til 60 mg/dag). Alle patienter modtog en standardiseret dosis af prednison 60 mg/dag ved studieindgang efterfulgt af en obligatorisk konisk tidsplan med komplet kortikosteroid -seponering inden uge 15.
Undersøgelse UV II evaluerede 226 patienter med inaktiv uveitis, mens de blev behandlet med kortikosteroider (oral prednison 10 til 35 mg/dag) ved baseline for at kontrollere deres sygdom. Patienter gennemgik efterfølgende en obligatorisk konisk tidsplan med komplet corticosteroid -seponering inden uge 19.
Klinisk respons
Resultater fra begge undersøgelser demonstrerede statistisk signifikant reduktion af risikoen for behandlingssvigt hos patienter behandlet med HUMIRA versus patienter, der fik placebo. I begge undersøgelser bidrog alle komponenter i det primære slutpunkt kumulativt til den overordnede forskel mellem Humira og placebogrupper (tabel 21).
Tabel 21: Tid til behandlingssvigt i undersøgelser UV I og UV II
| Uv i | Uv iI | |||||
| Placebo (N = 107) | Humira (N = 110) | HR [95% CI] a | Placebo (N = 111) | Humira (N = 115) | HR [95% CI] a | |
| Fiasko b n (%) | 84 (78.5) | 60 (NULL,5) | 0.50 [0,36 0,70] | 61 (55.0) | 45 (39.1) | 0.57 [0,39 0,84] |
| Median tid til fiasko (måneder) [95% CI] | 3.0 [2.7 3.7] | 5.6 [3.9 9.2] | N/a | 8.3 [4.8 12.0] | Ne c | N/a |
| a HR for Humira versus placebo fra proportional farer regression med behandling som faktor. b Behandlingssvigt ved eller efter uge 6 i undersøgelse UV I eller ved eller efter uge 2 i undersøgelse UV II blev talt som begivenhed. Personer, der ophørte med undersøgelsen, blev censureret på tidspunktet for dropping. c Ne = ikke estimable. Fewer than half of at-risk subjects had an event. |
Figur 3: Kaplan-Meier-kurver, der opsummerer tid til behandlingssvigt på eller efter uge 6 (undersøgelse UV I) eller uge 2 (undersøgelse UV II)
 |
Bemærk: s
Pædiatrisk uveitis
Sikkerheden og effektiviteten af Humira blev vurderet i en randomiseret dobbeltmasket placebokontrolleret undersøgelse af 90 pædiatriske patienter fra 2 til <18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Patients received either placebo or 20 mg Adalimumab (if < 30 kg) or 40 mg Adalimumab (if ≥ 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a mogatory røduction in topical corticosteroids within 3 months.
Det primære slutpunkt var ”tid til behandlingssvigt”. Kriterierne bestemmelse af behandlingssvigt blev forværret eller vedvarende ikke-forbedring i okulær betændelse eller forværring af okulære co-morbiditeter.
Klinisk respons
Humira significantly decreased the risk of treatment failure by 75% relative to placebo (HR = 0.25 [95% CI: 0.12 0.49]) (Table 22).
Tabel 22: analyseresultater af tid til behandlingssvigt (undersøgelse PUV-I)
| Placebo (N = 30) | Humira (N = 60) | HR (95% CI) a | |
| Fiasko (n[%]) | 18 (60%) | 16 (NULL,7%) | 0,25 (NULL,12 0,49) |
| Median tid til fiasko (uger) (95% CI) b | 24.1 (12.4 81.0) | NeC | |
| a HR af adalimumab versus placebo fra proportional farer regression med behandling som faktor. b Estimeret baseret på Kaplan-Meier-kurve. c Ne = ikke estimable. Fewer than half of at-risk subjects had an event. |
Figur 4: Kaplan-Meier-kurver, der opsummerer tid til behandlingssvigt (undersøgelse PUV-I)
 |
Undersøg puv-i
Bemærk: P = placebo (nummer i risiko); H = Humira (nummer i fare).
Referencer
1.National Cancer Institute. Overvågningsepidemiologi og slutresultat Database (SEER) -program. SEER-forekomst Råpriser 17 Registre 2000-2007.
Patientinformation til Humira
Humira ®
(Hu-Mare-ah)
(adalimumab)
injektion til subkutan brug
Læs medicinguiden, der følger med Humira, før du begynder at tage den, og hver gang du får en påfyldning. Der kan være nye oplysninger. Denne medicinguide indtager ikke stedet for at tale med din læge om din medicinske tilstand eller behandling.
Hvad er de vigtigste oplysninger, jeg skal vide om Humira?
Humira is a medicine that affects your immune system. Humira can lower the ability of your immune system to fight infections. Der er sket alvorlige infektioner hos mennesker, der tager Humira. Disse alvorlige infektioner inkluderer tuberkulose (TB) og infektioner forårsaget af vira svampe eller bakterier, der har spredt sig over hele kroppen. Nogle mennesker er døde af disse infektioner.
- Din læge skal teste dig for TB, inden du starter Humira.
- Din læge skal tjekke dig nøje for tegn og symptomer på TB under behandling med Humira.
Du skal ikke begynde at tage Humira, hvis du har nogen form for infektion, medmindre din læge siger, at det er okay.
Før du starter Humira, fortæl din læge, hvis du:
- Tror du har en infektion eller har symptomer på en infektion, såsom:
- Feber sved eller kulderystelser
- Muskelsmerter
- hoste
- åndenød
- Blod i slim
- varm rød eller smertefuld hud eller sår på din krop
- Diarré eller mavesmerter
- brænder, når du tisser eller tisser oftere end normalt
- Føl dig meget træt
- vægttab
- behandles for en infektion.
- Få en masse infektioner eller har infektioner, der fortsætter med at vende tilbage.
- har diabetes.
- har TB eller har været i tæt kontakt med nogen med TB.
- blev født i boede i eller rejste til lande, hvor der er mere risiko for at få TB. Spørg din læge, hvis du ikke er sikker.
- Live eller har boet i visse dele af landet (såsom Ohio og Mississippi River Valleys), hvor der er en øget risiko for at få visse former for svampeinfektioner (histoplasmosis coccidioidomycosis eller blastomycosis). Disse infektioner kan ske eller blive mere alvorlige, hvis du bruger Humira. Spørg din læge, hvis du ikke ved, om du har boet i et område, hvor disse infektioner er almindelige.
- har eller har haft hepatitis B.
- Brug medicinen orencia (abatacept) kineret (rituximab) eller purinetol (6-mercaptopurin 6-mp).
- er planlagt til at have større operation.
Efter at have startet Humira, skal du ringe til din læge med det samme Hvis du har en infektion eller et tegn på en infektion.
Humira can make you more likely to get infections or make any infection that you may have worse.
Kræft
- For børn og voksne, der tager tumor nekrose faktor (TNF) -klokkere inklusive Humira, kan chancerne for at få kræft stige.
- Der har været tilfælde af usædvanlige kræftformer hos børn teenagere og unge voksne, der bruger TNF-blokkere.
- Mennesker med reumatoid arthritis (RA), især mere alvorlig RA, kan have en større chance for at få en slags kræft kaldet lymfom.
- Hvis du bruger TNF -blokkeere inklusive Humira, kan din chance for at få to typer hudkræft stige (basalcellekræft og pladecellekræft i huden). Disse typer kræft er generelt ikke livstruende, hvis de behandles. Fortæl din læge, hvis du har et stød eller åbent ømhed, der ikke heles.
- Nogle mennesker, der modtog TNF-blokkeere, inklusive Humira, udviklede en sjælden type kræft kaldet hepatosplenisk T-celle lymfom. Denne type kræft resulterer ofte i død. De fleste af disse mennesker var mandlige teenagere eller unge mænd. De fleste mennesker blev også behandlet for Crohns sygdom eller ulcerøs colitis med en anden medicin kaldet Imuran (azathioprin) eller purinetol (6-mercaptopurin 6-MP).
Hvad er Humira?
Humira is a medicine called a Tumor Necrosis Factor (TNF) blocker. Humira is used:
- For at reducere tegn og symptomer på:
- Moderat til svær RA hos voksne. Humira can be used alone with methotrexate or with certain other medicines.
- Moderat til svær polyartikulær juvenil idiopatisk arthritis (JIA) hos børn 2 år og ældre. Humira kan bruges alene eller med methotrexat.
- Psoriasis arthritis (PSA) hos voksne. Humira can be used alone or with certain other medicines.
- Ankyloserende spondylitis (AS) hos voksne.
- Moderat til svær hidradenitis suppurativa (HS) hos mennesker 12 år og ældre.
- At behandle moderat til svær Crohns sygdom (CD) hos voksne og børn 6 år og ældre.
- At behandle moderat til svær ulcerøs colitis (UC) hos voksne og børn 5 år og ældre. Det vides ikke, om Humira er effektiv hos mennesker, der stoppede med at reagere på eller ikke kunne tolerere TNF-blokkeringsmediciner.
- At behandle moderat til svær kronisk (varer lang tid) plakspsoriasis (PS) hos voksne der har tilstanden i mange områder af deres krop, og som kan drage fordel af at tage injektioner eller piller (systemisk terapi) eller fototerapi (behandling ved hjælp af ultraviolet lys alene eller med piller).
- At behandle ikke-infektiøs mellemliggende posterior og panuveitis hos voksne og børn 2 år og ældre.
Hvad skal jeg fortælle min læge, før jeg tager Humira?
Humira may ikke be right for you. Before starteing Humira tell your doctor about all of your medical conditions including if you:
- har en infektion. Se Hvad er de vigtigste oplysninger, jeg skal vide om Humira?
- har eller har haft kræft.
- Har følelsesløshed eller prikken eller har en sygdom, der påvirker dit nervesystem, såsom multipel sklerose eller Guillain-Barré-syndrom.
- har eller havde hjertesvigt.
- har for nylig modtaget eller planlagt til at modtage en vaccine. Du modtager muligvis vacciner undtagen for levende vacciner, mens du bruger Humira. Børn skal bringes ajour med alle vacciner, inden de starter Humira.
- er allergiske over for gummi eller latex. Fortæl din læge, hvis du har allergi mod gummi eller latex.
- Nåledækslet for Humira Pen 40 mg/0,8 ml Humira 40 mg/0,8 ml Foretagret sprøjte Humira 20 mg/0,4 ml Foretagsprøjtning og Humira 10 mg/0,2 ml Præfyldt sprøjte kan indeholde naturgummi eller latex.
- The black needle cover for the HUMIRA Pen 80 mg/0.8 mL HUMIRA 80 mg/0.8 mL prefilled syringe HUMIRA Pen 40 mg/0.4 mL HUMIRA 40 mg/0.4 mL prefilled syringe HUMIRA 20 mg/0.2 mL prefilled syringe HUMIRA 10 mg/0.1 mL prefilled syringe and the vial stopper on the HUMIRA institutional use vial are Ikke lavet med naturgummi eller latex.
- er allergiske over for Humira eller over for nogen af dets ingredienser. Se slutningen på denne medicinvejledning for en liste over ingredienser i Humira.
- er gravide eller planlægger at blive gravid amning eller planlægge at amme. Du og din læge skal beslutte, om du skal tage Humira, mens du er gravid eller ammer.
- Få en baby, og du brugte Humira under din graviditet. Fortæl din babys læge, før din baby modtager vacciner.
Fortæl din læge om alle de medicin, du tager inklusive receptpligtige og over-counter medicin vitaminer og urtetilskud.
Fortæl især din læge, hvis du bruger:
- Orencia (Abatacept) Kineret (Anakinra) Remicade (Infliximab) Enbrel (Etanercept) Cimzia (certolizumab pegol) eller Simponi (golimumab), fordi du ikke skal bruge Humira, mens du også bruger en af disse medicin.
- Rituxan (rituximab). Din læge ønsker måske ikke at give dig Humira, hvis du for nylig har modtaget Rituxan (Rituximab).
- Imuran (azathioprin) eller purinetol (6-mercaptopurin 6-MP).
Opbevar en liste over dine medicin med dig for at vise din læge og farmaceut, hver gang du får en ny medicin.
Hvordan skal jeg tage Humira?
- Humira is given by an injection under the skin. Your doctor will tell you how often to take an injection of Humira. This is based on your condition to be treated. Injicer ikke Humira oftere, end du fik ordineret.
- Se instruktionerne til brug inde i kartonen for komplette instruktioner til den rigtige måde til at forberede og injicere Humira.
- Sørg for, at du har fået vist, hvordan du injicerer Humira, før du gør det selv. Du kan ringe til din læge eller 1-800-4Humira (1-800-448-6472), hvis du har spørgsmål om at give dig selv en injektion. En person, du kender, kan også hjælpe dig med din injektion, efter at de er blevet vist, hvordan du forbereder og injicerer Humira.
- Gør ikke Prøv at injicere Humira selv, indtil du har fået vist den rigtige måde at give injektionerne på. Hvis din læge beslutter, at du eller en plejeperson muligvis kan give dine injektioner af Humira derhjemme, skal du modtage træning på den rigtige måde til at forberede og injicere Humira.
- Gør ikke miss any doses of Humira unless your doctor says it is okay. If you forget to take Humira inject a dose as soon as you remember. Then take your next dose at your regular scheduled time. This will put you back on schedule. In case you are ikke sure when to inject Humira call your doctor or pharmacist.
- Hvis du tager mere Humira, end du fik at vide at tage din læge.
Hvad er de mulige bivirkninger af Humira?
Humira can cause serious side effects including:
Se hvad er de vigtigste oplysninger, jeg burde vide om Humira?
- Alvorlige infektioner.
Din læge vil undersøge dig for TB og udføre en test for at se, om du har TB. Hvis din læge føler, at du er i fare for TB, kan du blive behandlet med medicin mod TB, før du begynder behandling med Humira og under behandling med Humira. Selv hvis din TB -test er negativ, skal din læge omhyggeligt overvåge dig for TB -infektioner, mens du tager Humira. Mennesker, der havde en negativ TB -hudtest, før de modtog Humira, har udviklet aktiv TB. Fortæl din læge, hvis du har nogen af følgende symptomer, mens du tager eller efter at have taget Humira:
- hoste that does ikke go away
- Feber med lav kvalitet
- vægttab
- Tab af kropsfedt og muskler (spild)
- Hepatitis B -infektion hos mennesker, der bærer virussen i deres blod.
Hvis du er en bærer af hepatitis B -virus (en virus, der påvirker leveren), kan virussen blive aktiv, mens du bruger Humira. Din læge skal udføre blodprøver, før du starter behandling, mens du bruger Humira og i flere måneder efter du stopper behandlingen med Humira. Fortæl din læge, hvis du har nogen af følgende symptomer på en mulig hepatitis B -infektion:
- Muskelsmerter
- Føl dig meget træt
- Mørk urin
- Hud eller øjne ser gule ud
- lidt eller ingen appetit
- opkast
- Lerfarvede tarmbevægelser
- feber
- kulderystelser
- Mave ubehag
- Skinudslæt
- Allergiske reaktioner. Allergiske reaktioner kan ske hos mennesker, der bruger Humira. Ring til din læge eller få medicinsk hjælp med det samme, hvis du har nogen af disse symptomer på en alvorlig allergisk reaktion:
- elveblest
- problemer med at trække vejret
- Hævelse af dine ansigts øjne læber eller mund
- Problemer med nervesystemer. Tegn og symptomer på et nervesystemproblem inkluderer: følelsesløshed eller prikkende problemer med din synssvaghed i dine arme eller ben og svimmelhed.
- Blodproblemer. Din krop gør muligvis ikke nok af de blodlegemer, der hjælper med at bekæmpe infektioner eller hjælpe med at stoppe blødningen. Symptomerne inkluderer en feber, der ikke går væk med blå mærker eller blødning meget let eller ser meget bleg ud.
- Ny hjertesvigt eller forværring af hjertesvigt, du allerede har. Ring til din læge med det samme Hvis du får nye forværrede symptomer på hjertesvigt, mens du tager Humira, herunder:
- åndenød
- Pludselig vægtøgning
- Hævelse af dine ankler eller fødder
- Immunreaktioner inklusive et lupuslignende syndrom. Symptomerne inkluderer ubehag i brystet eller smerter, der ikke forsvinder med åndenødesmerter eller udslæt på dine kinder eller arme, der bliver værre i solen. Symptomerne kan forbedre sig, når du stopper Humira.
- Leverproblemer. Leverproblemer kan ske hos mennesker, der bruger TNF-blokkeringsmediciner. Disse problemer kan føre til leversvigt og død. Ring til din læge med det samme, hvis du har nogen af disse symptomer:
- Føl dig meget træt
- Dårlig appetit eller opkast
- Hud eller øjne ser gule ud
- Smerter på højre side af din mave (mave)
- Psoriasis. Nogle mennesker, der brugte Humira, havde ny psoriasis eller forværring af psoriasis, de allerede havde. Fortæl din læge, hvis du udvikler røde skællende pletter eller hævede buler, der er fyldt med pus. Din læge kan beslutte at stoppe din behandling med Humira.
Ring til din læge eller få medicinsk behandling med det samme, hvis du udvikler nogen af ovenstående symptomer. Din behandling med Humira kan stoppes.
De mest almindelige bivirkninger af Humira inkluderer:
- Reaktioner på injektionsstedet: Rødhed udslæt hævende kløe eller blå mærker. Disse symptomer forsvinder normalt inden for et par dage. Ring med din læge med det samme, hvis du har smerter i smerter eller hævelse omkring injektionsstedet, der ikke forsvinder inden for et par dage eller bliver værre.
- Øvre luftvejsinfektioner (inklusive sinusinfektioner).
- hovedpines.
- udslæt.
Dette er ikke alle de mulige bivirkninger med Humira. Fortæl din læge, hvis du har nogen bivirkning, der generer dig, eller som ikke forsvinder. Spørg din læge eller apotek for mere information.
Ring til din læge for medicinsk rådgivning om bivirkninger. Du kan rapportere bivirkninger til FDA på 1800- FDA-1088.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet ved 36 ° F til 46 ° F (2 ° C til 8 ° C). Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke Frys Humira. Gør ikke Brug Humira, hvis den er frosset, selvom det er optøet.
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -karton -dosisbakke eller forudfyldt sprøjte. Brug ikke Humira efter udløbsdatoen.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C (25 ° C) i op til 14 dage. Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Kast Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke blevet brugt inden for 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
- Gør ikke use a Pen or prefilled syringe if the liquid is cloudy discolorød or has flakes or particles in it.
- Gør ikke drop or crush Humira. The prefilled syringe is glass.
Hold Humira -injektionsartikler og alle andre medicin uden for børns rækkevidde.
Generel information om sikker og effektiv anvendelse af Humira.
Medicin er undertiden ordineret til andre formål end dem, der er anført i en medicinguide. Brug ikke Humira til en betingelse, som den ikke blev ordineret til. Giv ikke Humira til andre mennesker, selvom de har den samme betingelse. Det kan skade dem. Denne medicinguide opsummerer de vigtigste oplysninger om Humira. Hvis du gerne vil have flere oplysninger, skal du tale med din læge. Du kan bede din farmaceut eller læge om information om Humira, der er skrevet til sundhedsfagfolk.
Hvad er ingredienserne i Humira?
Aktiv ingrediens: Adalimumab
Humira Pen 40 mg/0.8 mL Humira 40 mg/0.8 mL prefilled syringe Humira 20 mg/0.4 mL prefilled syringe Humira 10 mg/0.2 mL prefilled syringe og Humira 40 mg/0.8 mL institutional use vial:
Inaktive ingredienser: Citronsyre monohydratdibasisk natriumphosphatdihydrat mannitol monobasisk natriumphosphatdihydrat polysorbat 80 natriumchlorid natriumcitrat og vand til injektion. Natriumhydroxid tilsættes som nødvendigt for at justere pH.
Humira Pen 80 mg/0.8 mL Humira 80 mg/0.8 mL prefilled syringe Humira Pen 40 mg/0,4 ml Humira 40 mg/0,4 ml prefilled syringe Humira 20 mg/0.2 mL prefilled syringe og Humira 10 mg/0.1 mL prefilled syringe:
Inaktive ingredienser: Mannitol polysorbat 80 og vand til injektion.
Brug til brug
Humira ®
(Hu-Mare-ah)
(adalimumab)
40 mg/0,8 ml
Enkeltdosis pen
Gør ikke Prøv at injicere Humira selv, indtil du har fået vist den rigtige måde at give injektionerne og have læst og forstå disse brugsinstruktioner. Hvis din læge beslutter, at du eller en plejeperson muligvis kan give dine injektioner af Humira derhjemme, skal du modtage træning på den rigtige måde til at forberede og injicere Humira. Det er vigtigt, at du læser forstå og følger disse instruktioner, så du injicerer Humira på den rigtige måde. Det er også vigtigt at tale med din læge for at være sikker på, at du forstår dine Humira -doseringsinstruktioner. For at hjælpe dig med at huske, hvornår du skal injicere Humira, kan du markere din kalender på forhånd. Ring til din sundhedsudbyder, hvis du eller din plejeperson har spørgsmål om den rigtige måde at injicere Humira på.
VIGTIG:
- Gør ikke Brug Humira, hvis den er frosset, selvom det er optøet.
- Humira -penen indeholder glas. Slip ikke eller knus pennen, fordi glasset inde kan gå i stykker.
- Hver Humira -pen har 2 kasketter på. Fjern ikke den grå hætte (cap
- Når den blommerfarvede knap på Humira-pennen presses for at give din dosis Humira, vil du høre en høj kliklyd.
- Du skal øve dig på at injicere Humira med din læge eller sygeplejerske, så du ikke er forskrækket af dette klik, når du begynder at give dig selv injektionerne derhjemme.
- Den høje kliklyd betyder starten af injektionen.
- Du vil vide, at injektionen er færdig, når den gule indikator vises fuldt ud i vinduesvisningen og holder op med at bevæge sig.
Se afsnittet nedenfor Forberedelse af Humira -pen.
Saml forsyningerne til din injektion
- Du har brug for følgende forsyninger til hver injektion af Humira.
Find en ren flad overflade til at placere forsyningerne på.
- 1 alkoholpind
- 1 bomuldskugle eller gasbindel (ikke inkluderet i din Humira -karton)
- 1 Humira Pen (se Figur a )
- Punkteringsbestandig Sharps bortskaffelsesbeholder til Humira Pen bortskaffelse (ikke inkluderet i din Humira-karton). Se Hvordan skal jeg smide (bortskaffe) den brugte Humira -pen? Afsnit i slutningen af denne brugsanvisning.
Hvis mere komfortabel, tag din Humira -pen ud af køleskabet 15 til 30 minutter inden injektion for at lade væsken nå stuetemperatur. Gør ikke Fjern den grå hætte (cap Gør ikke Varm Humira på nogen anden måde (for eksempel Gør ikke Varm det i en mikrobølgeovn eller i varmt vand).
Hvis du ikke har alle de forsyninger, skal du give dig selv en injektion, gå til et apotek eller ringe til din farmaceut. Figuren nedenfor viser, hvordan Humira -penen ser ud. Se Figur a .
Figur a
 |
Kontroller kartondosisbakken og Humira -pen.
Se Hvordan skal jeg opbevare Humira? Afsnit i slutningen af denne brugsanvisning.
Figur b
 |
Figur c
 |
Vælg injektionsstedet
Forbered injektionsstedet
Forberedelse af Humira -pen
Figur e
 |
Placer pennen og injicer Humira
Figur j
 |
- Sørg for, at navnet Humira vises på kartondosisbakken og Humira pen -etiket.
- Gør ikke use og Ring til Din læge eller farmaceut hvis:
- Du slipper eller knuser din Humira -pen.
- Forseglingerne på toppen eller bunden af kartonen er brudt eller mangler.
- Udløbsdatoen på kartondosisbakken og pen er gået.
- Humira -penen er frosset eller efterladt i direkte sollys.
- Humira has been kept at room temperature for longer than 14 Dage eller Humira er blevet opbevaret over 25 ° C (25 ° C).
- Hold pennen med den grå hætte (hætte
- Sørg for, at mængden af væske i pennen er på fyldlinjen eller tæt på fyldlinjen set gennem vinduet. Dette er den fulde dosis af Humira, som du vil injicere. Se Figur b .
- Hvis pennen ikke har den fulde mængde væske Gør ikke use that Pen. Ring til din farmaceut.
- Vend pennen og hold pennen med den grå hætte (hætte Figur c .
- Kontroller opløsningen gennem vinduerne på siden af pennen for at sikre, at væsken er klar og farveløs. Gør ikke use Din Humira -pen, hvis væsken er overskyet misfarvet, eller hvis den har flager eller partikler i den. Ring til din farmaceut. Det er normalt at se en eller flere bobler i vinduet.
- Vask og tør dine hænder godt.
- Vælg et injektionssted på:
Figur d
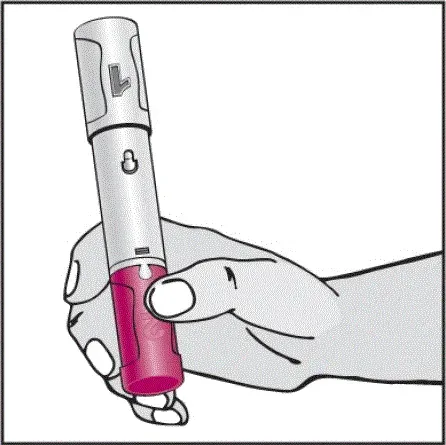
- forsiden af dine lår eller
- din underliv (mave). Hvis du vælger dit mave, skal du ikke bruge området 2 tommer omkring din maveknap (navle). Se Figur d .
- Vælg et andet sted, hver gang du giver dig selv en injektion. Hver ny injektion skal gives mindst en tomme fra et sted, du brugte før.
- Gør ikke Injicere Humira i huden, det er:
- øm (øm)
- forslået
- rød
- hård
- arret eller hvor du har strækmærker
- Hvis du har psoriasis Gør ikke Injicere direkte i enhver hævet tyk rød eller skællende hudplaster eller læsioner på din hud.
- Gør ikke inject through your clothes.
- Tør injektionsstedet med en alkoholforberedelse (swab) ved hjælp af en cirkulær bevægelse.
- Gør ikke Rør ved dette område igen, før du giver injektionen. Lad huden tørre inden injektion. Gør ikke Ventilator eller blæse på det rene område.
- Gør ikke remove the gray cap (Cap # 1) or the plum-colorød cap (Cap # 2) until right before your injection.
- Hold midten af pennen (grå krop) med den ene hånd, så du ikke rører ved den grå hætte (hætte Figur e .
- Med din anden hånd træk den grå hætte (cap Figur f .
- Smid den grå hætte (cap væk
Figur f
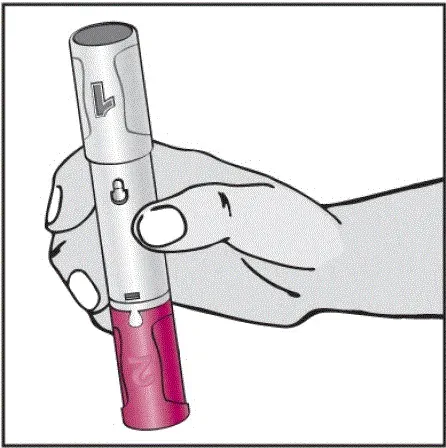
- Gør ikke Sæt den grå hætte (cap
- Den hvide nåleskærm, der dækker nålen, kan nu ses.
- Gør ikke Rør nålen med fingrene, eller lad nålen røre ved noget.
- Du kan se et par dråber væske komme ud af nålen. Dette er normalt.
- Fjern den blommerfarvede hætte (cap
Den blommefarvede aktivatorknap:
Figur g

Figur h
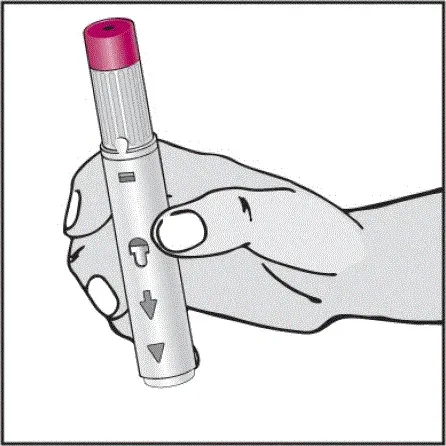
- Gør ikke put the plum-colorød cap (Cap # 2) back on pennen because it could cause medicine to come out of the syringe.
- Drej pennen, så den blomme-farvede aktivator-knap er påpeget. Se Figur g .
- Gør ikke Tryk på den blomme-farvede aktivatorknap, indtil du er klar til at injicere Humira. Ved at trykke på den blommefarvede aktivatorknap frigiver medicinen fra pennen.
- Hold pennen, så du kan se vinduet. Se Figur h . Det er normalt at se en eller flere bobler i vinduet.
- Placer pennen:
Figur i
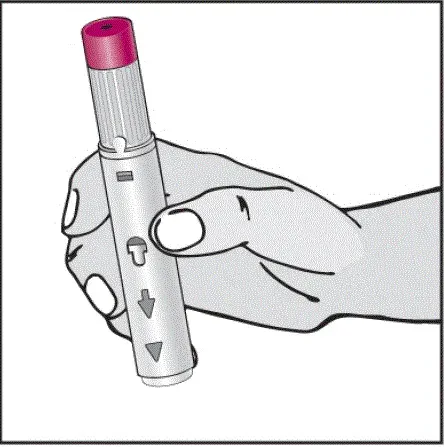
- Presse Området med den rensede hud og holder den fast, indtil injektionen er afsluttet. Se Figur i . Du vil injicere i dette hævede hudområde.
- Placer den hvide ende af pennen lige (i en vinkel på 90 °) og flat against the raised area of your skin that you are squeezing. Placere pennen so that it will ikke inject the needle into your fingers that are holding the raised skin. Se Figur j .
- Injicer Humira
Figur k
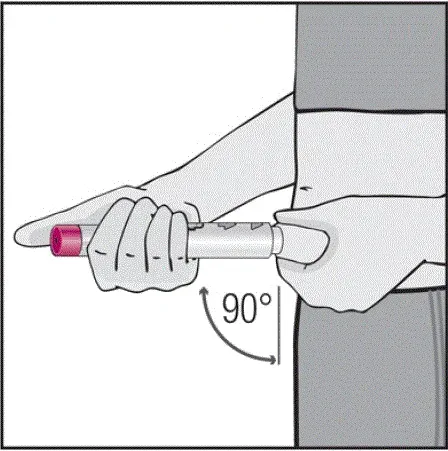
Figur l
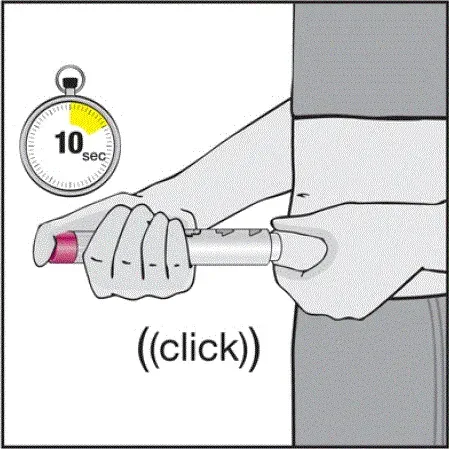
- Det er vigtigt, at du trækker pennen fast ned Hele vejen mod injektionsstedet inden injektionen startes.
- Bliv ved med at skubbe ned For at forhindre, at pennen bevæger sig væk fra huden under injektionen.
- Tryk på den blommefarvede aktivatorknap med tommelfingeren for at begynde injektionen. Prøv ikke at dække vinduet. Se Figur k .
- Du vil høre et højt 'klik', når du trykker på den blomme-farvede aktivatorknap. Det høje klik betyder starten af injektionen.
- Fortsæt med at trykke på den blommefarvede aktivatorknap, og fortsæt med at skubbe pennen mod din klemte hævede hud, indtil al medicinen er injiceret. Dette kan tage op til 10 sekunder, så tæl langsomt til ti. Bliv ved med at skubbe pennen mod det pressede hævede hud på dit injektionssted hele tiden, så du får den fulde dosis medicin.
- Du vil vide, at injektionen er færdig, når den gule indikator fuldt ud vises i vinduesvisningen og holder op med at bevæge sig. Se Figur l .
- Når injektionen er færdig, skal du langsomt trække pennen fra din hud. Den hvide nåleskærm bevæger sig for at dække nålspidsen. Se Figur m .
Figur m
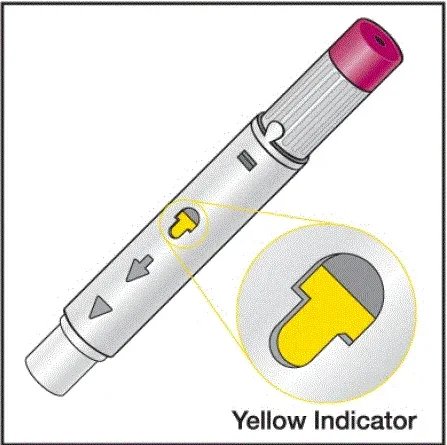
- Gør ikke touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Der kan være en lille mængde væske på injektionsstedet. Dette er normalt.
- Tryk på en bomuldskugle eller gasbind over injektionsstedet og hold den i 10 sekunder. Gør ikke Gnid injektionsstedet. Du har muligvis let blødning. Dette er normalt.
- Kast væk (bortskaffer) din brugte Humira -pen i en skarp bortskaffelsescontainer med det samme efter brug. Se afsnittet Hvordan skal jeg bortskaffe den brugte Humira -pen?
- Hold en oversigt over datoer og placering af dine injektionssteder. For at hjælpe dig med at huske, hvornår du skal tage Humira, kan du markere din kalender på forhånd.








Hvordan skal jeg smide (bortskaffe) den brugte Humira -pen?
Figur n
 |
- Sæt din pen i en FDA-klaret skarp bortskaffelsescontainer med det samme efter brug. Se Figur n . Gør ikke throw away pennen in your household tudslæt.
- Gør ikke try to touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Hvis du ikke har en FDA-Cleared Sharps bortskaffelsesbeholder, kan du bruge en husholdningsbeholder, der er:
- lavet af en kraftig plastik
- kan lukkes med et tæt passende punkteringsbestandigt låg uden at skarpe er i stand til at komme ud
- lodret og stabil under brug
- lækagebestandig og
- Korrekt mærket til at advare om farligt affald inde i beholderen.
- Når din Sharps -bortskaffelsescontainer er næsten fuld, skal du følge dine samfundsretningslinjer for den rigtige måde at bortskaffe din Sharps Disposal Container. Der kan være statslige eller lokale love om, hvordan du skal smide brugte nåle og sprøjter. For mere information om Safe Sharps bortskaffelse og for specifik information om Sharps bortskaffelse i den stat, du bor i, skal du gå til FDA's websted på: https://www.fda.gov/safesharpsdisposal.
- For sikkerheden og sundheden for dig og andre genanvendes aldrig dine Humira-penne igen.
- De brugte alkoholpuder bomuldskugler dosisbakker og emballage kan placeres i dit husholdningspass.
- Gør ikke dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Gør ikke recycle your used sharps disposal container.
- Hold altid skarpe beholder uden for børns rækkevidde.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet mellem 36 ° F (2 ° C (2 ° C (2 ° C). Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke Frys Humira. Gør ikke Brug Humira, hvis den er frosset, selvom det er optøet.
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -kartondosisbakken eller pen. Gør ikke Brug Humira efter udløbsdatoen.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C 14 dage. Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Smid Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke blevet brugt indeni 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
- Gør ikke use a Pen if the liquid is cloudy discolorød or has flakes or particles in it.
- Gør ikke drop or crush Humira.
- Hold Humira -injektionsartikler og alle andre medicin uden for børns rækkevidde.
Brug til brug
Humira ®
(Hu-Mare-ah)
(adalimumab)
40 mg/0,4 ml
Enkeltdosis pen
Før du injicerer: Din sundhedsudbyder skal vise dig, hvordan du bruger Humira, før du bruger den for første gang. Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Hvis du har brug for hjælp.
Figur a: Humira Enkeltdosis pen
Hvad gør 800 mg ibuprofen
 |
Vigtige oplysninger, du har brug for at vide, før du injicerer Humira
Gør ikke Brug pennen og ring til din sundhedsudbyder eller farmaceut, hvis:
- Væske er overskyet misfarvet eller har flager eller partikler i det
- Udløbsdato er gået
- Væske er frosset (selvom tøet) eller efterladt i direkte sollys
- Pennen er droppet eller knust
Hold hætterne på indtil lige før din injektion.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet mellem 36 ° F (2 ° C (2 ° C (2 ° C).
- Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke freeze
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -kartondosisbakken eller pen.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C 14 dage.
- Smid Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke brugt indeni 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
Hold Humira -injektionsartikler og alle andre medicin uden for børn.
Læs instruktioner på alle sider, før du bruger Humira -pen
Tage Humira out of the refrigerator.
Forlade Humira at room temperature for 15 til 30 minutter inden injektion.
- Gør ikke Fjern den grå hætte (cap
- Gør ikke Varm Humira på nogen anden måde. For eksempel Gør ikke Varm det i en mikrobølgeovn eller i varmt vand.
- Gør ikke Brug pennen, hvis væske er frosset (selvom tøet)
 |
Check Udløbsdato på penetiketten. Gør ikke Brug pennen, hvis udløbsdatoen er gået.
Placere Følgende på en ren flad overflade:
- 1 enkeltdosis pen og alkoholpinde
- 1 bomuldskugle eller gasbindel (ikke inkluderet)
- Punkteringsbestandig Sharps bortskaffelsesbeholder (ikke inkluderet). Se trin 9 i slutningen af denne instruktioner til brug til instruktioner om, hvordan man smider væk (bortskaffer) din Humira -pen
Vask og tør dine hænder.
 |
Vælge Et injektionssted:
- På fronten af dine lår eller
- Dit mave (mave) mindst 2 inches fra din navle (maveknap)
- Forskellig fra dit sidste injektionssted
Tørre Injektionsstedet i en cirkulær bevægelse med alkoholpinden.
- Gør ikke Injekt gennem tøj
- Gør ikke Injicer i huden, der er øm mærket rød hård arret har strækmærker eller områder med psoriasis plaques
 |
Holde pennen med den grå hætte Check Vinduet.
- Det er normalt at se 1 eller flere bobler i vinduet
- Sørg for, at væsken er klar og farveløs
- Gør ikke Brug pennen, hvis væsken er overskyet misfarvet eller har flager eller partikler i den
- Gør ikke Brug pennen, hvis den er droppet eller knust
 |
Træk Den grå hætte
Kast hætten væk.
- Det er normalt at se et par dråber væske komme ud af nålen
Træk Den blommefarvede hætte
Kast hætten væk.
Drej pennen, så den hvide pil peger mod injektionsstedet.
 |
Presse Huden på dit injektionssted for at lave et hævet område og holde det fast, indtil injektionen er afsluttet.
Punkt Den hvide pil mod injektionsstedet.
Placere den hvide nåleskærm lige (90 ° vinkel) mod injektionsstedet.
Holde pennen, så du kan se inspektionsvinduet.
Gør ikke Tryk på knappen Plum Activator, indtil du er klar til at injicere.
 |
Det er vigtigt, at du trækker pennen fast ned Hele vejen mod injektionsstedet inden injektionen startes.
Bliv ved med at skubbe ned For at forhindre, at pennen bevæger sig væk fra huden under injektionen.
Trykke Blomme -aktivatorknappen og tæller langsomt for 10 sekunder.
- Et højt klik vil signalere starte af injektionen
- Bliv ved med at skubbe pennen ned fast mod injektionsstedet, indtil injektionen er afsluttet
- Injektion er afsluttet, når den gule indikator er stoppet med at bevæge sig
 |
Når injektionen afsluttes langsomt, skal du trække pennen fra huden. Den hvide nåleskærm dækker nålspidsen.
- En lille mængde væske på injektionsstedet er normalt
Hvis der er mere end et par dråber væske på injektionsstedets opkald 1-800-4Humira (1-800448- 6472) for hjælp.
Efter at have afsluttet injektionsstedet placerer en bomuldskugle eller gasbind på huden på injektionsstedet.
- Gør ikke gnide
- Let blødning på injektionsstedet er normalt
 |
Hvordan skal jeg bortskaffe den brugte Humira -pen?
- Sæt dine brugte nålepenne og skarpe i en FDA ryddet Sharps bortskaffelsescontainer med det samme efter brug. Gør ikke throw away (dispose of) loose needles syringes og pennen in the household tudslæt.
- Hvis du ikke har en FDA -ryddet Sharps bortskaffelsesbeholder, kan du bruge en husholdningsbeholder, der er:
- lavet af en kraftig plastik
- kan lukkes med et tæt passende punkteringsbestandigt låg uden at skarpe er i stand til at komme ud
- lodret og stabil under brug
- lækagebestandig og
- Korrekt mærket til at advare om farligt affald inde i beholderen.
- Når din Sharps -bortskaffelsescontainer er næsten fuld, skal du følge dine samfundsretningslinjer for den rigtige måde at bortskaffe din Sharps Disposal Container. Der kan være statslige eller lokale love om, hvordan du skal smide brugte nåle og sprøjter. For mere information om Safe Sharps bortskaffelse og for specifik information om Sharps bortskaffelse i den stat, du bor i, skal du gå til FDA's websted på: https://www.fda.gov/safesharpsdisposal.
- Gør ikke dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Gør ikke recycle your used sharps disposal container.
 |
Penen hætter alkoholpusboldbold eller gasbindedosisbakke og emballage kan placeres i dit husholdningspass.
Spørgsmål om at bruge Humira -pen
Hvad hvis jeg ikke har modtaget personligt uddannelse fra en sundhedsudbyder?
- Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Eller besøg www.humira.com, hvis du har brug for hjælp
Hvordan ved jeg, hvornår injektionen er færdig?
- Den gule indikator er stoppet med at bevæge sig. Dette tager op til 10 sekunder.
Hvad skal jeg gøre, hvis der er mere end et par dråber væske på injektionsstedet?
- Ring 1-800-4Humira (1-800-448-6472) for hjælp
Hvad hvis jeg ikke har en FDA-Cleared Sharps bortskaffelsesbeholder eller korrekt husholdningsbeholder?
- Ring 1-800-4Humira (1-800-448-6472) For en gratis FDA-klaret skarp bortskaffelsesbeholder
Altid Hold pennen og Sharps bortskaffelsesbeholder uden for børns rækkevidde.
Hold en oversigt over datoer og placeringer af dine injektioner. For at hjælpe med at huske, hvornår du skal tage Humira til at markere din kalender i forvejen.
 |
 |
Brug til brug
Humira ®
(Hu-Mare-ah)
(adalimumab)
Pakker indeholdende 80 mg/0,8 ml
Enkeltdosis pen
Før du injicerer: Din sundhedsudbyder skal vise dig, hvordan du bruger Humira, før du bruger den for første gang. Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Hvis du har brug for hjælp.
Figur a: Humira Enkeltdosis pen
 |
Vigtige oplysninger, du har brug for at vide, før du injicerer Humira
Gør ikke Brug pennen og ring til din sundhedsudbyder eller farmaceut, hvis:
- Væske er overskyet misfarvet eller har flager eller partikler i det
- Udløbsdato er gået
- Væske er frosset (selvom tøet) eller efterladt i direkte sollys
- Pennen er droppet eller knust
Hold hætterne på indtil lige før din injektion.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet mellem 36 ° F (2 ° C (2 ° C (2 ° C).
- Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke freeze
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -kartondosisbakken eller pen.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C 14 dage.
- Smid Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke brugt indeni 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
Hold Humira -injektionsartikler og alle andre medicin uden for børn.
Læs instruktioner på alle sider, før du bruger Humira -pen
Tage Humira out of the refrigerator.
Forlade Humira at room temperature for 15 til 30 minutter inden injektion.
- Gør ikke Fjern den grå hætte (cap
- Gør ikke Varm Humira på nogen anden måde. For eksempel Gør ikke Varm det i en mikrobølgeovn eller i varmt vand
- Gør ikke Brug pennen, hvis væske er frosset (selvom tøet)
 |
Check Udløbsdato på penetiketten. Gør ikke Brug pennen, hvis udløbsdatoen er gået.
Placere Følgende på en ren flad overflade:
- 1 enkeltdosis pen og alkoholpinde
- 1 bomuldskugle eller gasbindel (ikke inkluderet)
- Punkteringsbestandig Sharps bortskaffelsesbeholder (ikke inkluderet). Se trin 9 i slutningen af denne instruktioner til brug til instruktioner om, hvordan man smider væk (bortskaffer) din Humira -pen
Vask og tør dine hænder.
 |
Vælge Et injektionssted:
- På fronten af dine lår eller
- Dit mave (mave) mindst 2 inches fra din navle (maveknap)
- Forskellig fra dit sidste injektionssted
Tørre Injektionsstedet i en cirkulær bevægelse med alkoholpinden.
- Gør ikke Injekt gennem tøj
- Gør ikke Injicer i huden, der er øm mærket rød hård arret har strækmærker eller områder med psoriasis plaques
 |
Holde pennen med den grå hætte Check Vinduet.
- Det er normalt at se 1 eller flere bobler i vinduet
- Sørg for, at væsken er klar og farveløs
- Gør ikke Brug pennen, hvis væsken er overskyet misfarvet eller har flager eller partikler i den
- Gør ikke Brug pennen, hvis den er droppet eller knust
 |
Træk Den grå hætte
Kast hætten væk.
- Det er normalt at se et par dråber væske komme ud af nålen
Træk Den blommefarvede hætte
Kast hætten væk.
Tur pennen so that the white arrow points toward the injection site.
 |
Presse Huden på dit injektionssted for at lave et hævet område og holde det fast, indtil injektionen er afsluttet.
Punkt Den hvide pil mod injektionsstedet.
Placere den hvide nåleskærm lige (90 ° vinkel) mod injektionsstedet.
Holde pennen, så du kan se inspektionsvinduet.
 |
Gør ikke Tryk på knappen Plum Activator, indtil du er klar til at injicere.
Det er vigtigt, at du trækker pennen fast ned Hele vejen mod injektionsstedet inden injektionen startes.
Trykke Blomme -aktivatorknappen og tæller langsomt for 15 sekunder.
- Et højt klik vil signalere starte af injektionen
- Bliv ved med at skubbe pennen ned fast mod injektionsstedet, indtil injektionen er afsluttet
- Injektion er afsluttet, når den gule indikator er stoppet med at bevæge sig
 |
Når injektionen afsluttes langsomt, skal du trække pennen fra huden. Den hvide nåleskærm dækker nålspidsen.
- En lille mængde væske på injektionsstedet er normalt
Hvis der er mere end et par dråber væske på injektionsstedets opkald 1-800-4Humira (1-800448- 6472) for hjælp.
Efter at have afsluttet injektionsstedet placerer en bomuldskugle eller gasbind på huden på injektionsstedet.
- Gør ikke gnide
- Let blødning på injektionsstedet er normalt
 |
Hvordan skal jeg bortskaffe den brugte Humira -pen?
- Sæt dine brugte nålepenne og skarpe i en FDA ryddet Sharps bortskaffelsescontainer med det samme efter brug. Gør ikke throw away (dispose of) loose needles syringes og pennen in the household tudslæt.
- Hvis du ikke har en FDA-Cleared Sharps bortskaffelsesbeholder, kan du bruge en husholdningsbeholder, der er:
- lavet af en kraftig plastik
- kan lukkes med et tæt passende punkteringsbestandigt låg uden at skarpe er i stand til at komme ud
- lodret og stabil under brug
- lækagebestandig og
- Korrekt mærket til at advare om farligt affald inde i beholderen.
- Når din Sharps -bortskaffelsescontainer er næsten fuld, skal du følge dine samfundsretningslinjer for den rigtige måde at bortskaffe din Sharps Disposal Container. Der kan være statslige eller lokale love om, hvordan du skal smide brugte nåle og sprøjter. For mere information om Safe Sharps bortskaffelse og for specifik information om Sharps bortskaffelse i den stat, du bor i, skal du gå til FDA's websted på: https://www.fda.gov/safesharpsdisposal.
- Gør ikke dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Gør ikke recycle your used sharps disposal container.
 |
Penen hætter alkoholpusboldbold eller gasbindedosisbakke og emballage kan placeres i dit husholdningspass.
Spørgsmål om at bruge Humira -pen
Hvad hvis jeg ikke har modtaget en personuddannelse fra en sundhedsudbyder?
- Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Eller besøg www.humira.com, hvis du har brug for hjælp
Hvordan ved jeg, hvornår injektionen er færdig?
- Den gule indikator er stoppet med at bevæge sig. Dette tager op til 15 sekunder.
Hvad skal jeg gøre, hvis der er mere end et par dråber væske på injektionsstedet?
- Ring 1-800-4Humira (1-800-448-6472) for hjælp
Hvad hvis jeg ikke har en FDA-Cleared Sharps bortskaffelsesbeholder eller korrekt husholdningsbeholder?
- Ring 1-800-4Humira (1-800-448-6472) For en gratis FDA-klaret skarp bortskaffelsesbeholder
Altid Hold pennen og Sharps bortskaffelsesbeholder uden for børns rækkevidde.
Hold en oversigt over datoer og placeringer af dine injektioner. For at hjælpe med at huske, hvornår du skal tage Humira til at markere din kalender i forvejen.
 |
 |
Brug til brug
Humira ®
(Hu-Mare-ah)
(adalimumab)
40 mg/0,8 ml 20 MG/0.4 ML AND 10 MG/0.2 ML
Enkeltdosis forfyldt sprøjte
Gør ikke Prøv at injicere Humira selv, indtil du har fået vist den rigtige måde at give injektionerne og have læst og forstå disse brugsinstruktioner. Hvis din læge beslutter, at du eller en plejeperson muligvis kan give dine injektioner af Humira derhjemme, skal du modtage træning på den rigtige måde til at forberede og injicere Humira. Det er vigtigt, at du læser forstå og følger disse instruktioner, så du injicerer Humira på den rigtige måde. Det er også vigtigt at tale med din læge for at være sikker på, at du forstår dine Humira -doseringsinstruktioner. For at hjælpe dig med at huske, hvornår du skal injicere Humira, kan du markere din kalender på forhånd. Ring til din sundhedsudbyder, hvis du eller din plejeperson har spørgsmål om den rigtige måde at injicere Humira på.
Saml forsyningerne til din injektion
- Du har brug for følgende forsyninger til hver injektion af Humira.
Find en ren flad overflade til at placere forsyningerne på.
- 1 alkoholpind
- 1 bomuldskugle eller gasbindel (ikke inkluderet i din Humira -karton)
- 1 Humira -præfyldt sprøjte (se Figur a )
- Punkteringsbestandig Sharps bortskaffelsesbeholder til Humira-præfyldt bortskaffelse af sprøjte (ikke inkluderet i din Humira-karton). Se Hvordan skal jeg smide (bortskaffe) de brugte præfyldte sprøjter og nåle? Afsnit i slutningen af denne brugsanvisning.
Hvis mere komfortabel, skal du tage din Humira -præfyldte sprøjte ud af køleskabet 15 til 30 minutter inden injektion for at lade væsken nå stuetemperatur. Gør ikke Fjern nåledækslet, mens det kan nå ud til stuetemperatur. Gør ikke Varm Humira på nogen anden måde (for eksempel Gør ikke Varm det i en mikrobølgeovn eller i varmt vand).
Hvis du ikke har alle de forsyninger, skal du give dig selv en injektion, gå til et apotek eller ringe til din farmaceut.
Figuren nedenfor viser, hvordan en forudfyldt sprøjte ser ud. Se Figur a .
Figur a
 |
Check the carton dose tray og prefilled syringe
Se Hvordan skal jeg opbevare Humira? Afsnit i slutningen af denne brugsanvisning.
Vælg injektionsstedet
Forbered injektionsstedet
Gør ikke Ventilator eller blæse på det rene område.
Forbered sprøjten og nålen
Figur d
 |
Placer den forudfyldte sprøjte og injicer Humira
Placer sprøjten
Figur h
 |
Injicer Humira
- Sørg for, at navnet Humira vises på dosisbakken og forudfyldt sprøjte etiket.
- Gør ikke use og Ring til Din læge eller farmaceut hvis:
- Forseglingerne på toppen eller bunden af kartonen er brudt eller mangler.
- Humira -mærkningen har en udløbet dato. Kontroller udløbsdatoen på din Humira -karton og Gør ikke Brug, hvis datoen er gået.
- Den forudfyldte sprøjte er frosset eller efterladt i direkte sollys.
- Humira has been kept at room temperature for longer than 14 Dage eller Humira er blevet opbevaret over 25 ° C (25 ° C).
- Væsken i den forudfyldte sprøjte er overskyet misfarvet eller har flager eller partikler i den. Sørg for, at væsken er klar og farveløs.
- Vask og tør dine hænder godt.
- Vælg et injektionssted på:
Figur b
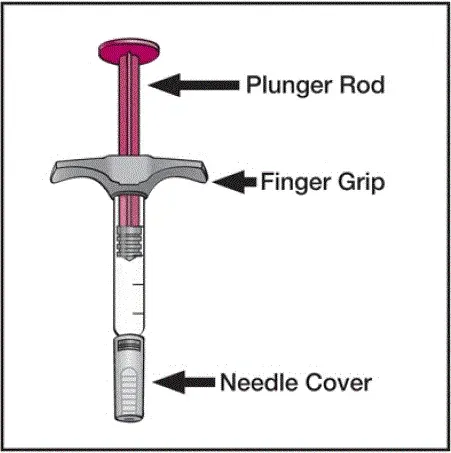
- forsiden af dine lår eller
- din underliv (mave). Hvis du vælger dit mave, skal du ikke bruge området 2 tommer omkring din maveknap (navle). Se Figur b .
- Vælg et andet sted, hver gang du giver dig selv en injektion. Hver ny injektion skal gives mindst en tomme fra et sted, du brugte før.
- Gør ikke Injicer i huden, det er:
- øm (øm)
- forslået
- rød
- hård
- arret eller hvor du har strækmærker
- Hvis du har psoriasis Gør ikke Injicere direkte i enhver hævet tyk rød eller skællende hudplaster eller læsioner på din hud.
- Gør ikke inject through your clothes.
- Tør injektionsstedet med en alkoholforberedelse (swab) ved hjælp af en cirkulær bevægelse.
- Gør ikke Rør ved dette område igen, før du giver injektionen. Lad huden tørre inden injektion.
- Check the fluid level in the syringe:
Figur c
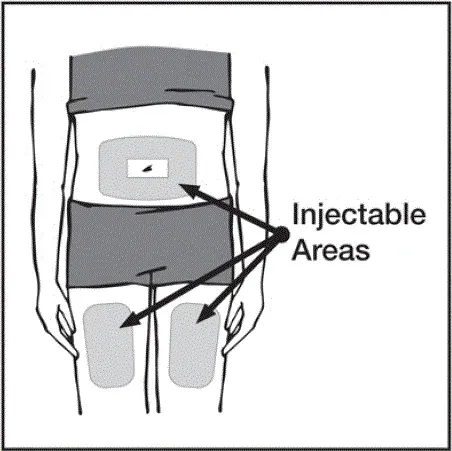
- Holde the syringe with the coverød needle pointing down. Se Figur c .
- Holde the syringe at eye level. Look closely to make sure that the amount of liquid in the syringe is the same or close to the:
- 0,8 ml linje for den 40 mg præfyldte sprøjte. Se Figur d .
- 0,4 ml linje for den 20 mg forudfyldte sprøjte. Se Figur d .
- 0,2 ml linje for den 10 mg forudfyldte sprøjte. Se Figur d .
- Toppen af væsken kan være buet. Hvis sprøjten ikke har den rigtige mængde væske Gør ikke use that syringe. Ring til din farmaceut.
- Fjern nåledækslet:
Figur e
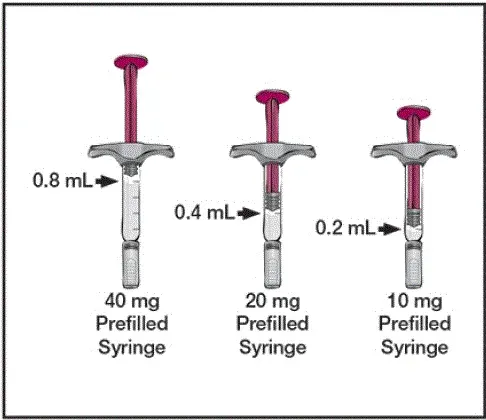
- Holde the syringe in one hog. With the other hog gently remove the needle cover. Se Figur e .
- Kast nåledækslet væk.
- Gør ikke Rør nålen med fingrene, eller lad nålen røre ved noget.
- Tur the syringe so the needle is facing up og hold the syringe at eye level with one hog so you can see the air in the syringe. Brug af din anden hånd Skub langsomt Stemplet ind for at skubbe luften ud gennem nålen. Se Figur f .
Figur f
- Du kan se en dråbe væske i slutningen af nålen. Dette er normalt.
- Holde the body of the prefilled syringe in one hog between the thumb og index finger. Holde the syringe in your hog like a pencil. Se Figur g .
Figur g
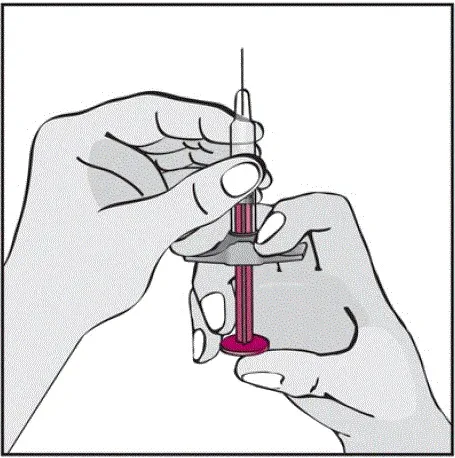
- Gør ikke Træk tilbage på stemplet når som helst.
- Med din anden hånd press forsigtigt området af den rensede hud og hold det fast. Se Figur h .
- Brug en hurtig dartlignende bevægelse indsæt nålen i den klemte hud omkring en 45 graders vinkel. Se Figur i .
Figur i

- Efter nålen er i slip af huden. Træk forsigtigt tilbage på stemplet.
Hvis blod vises i sprøjten:
Figur j
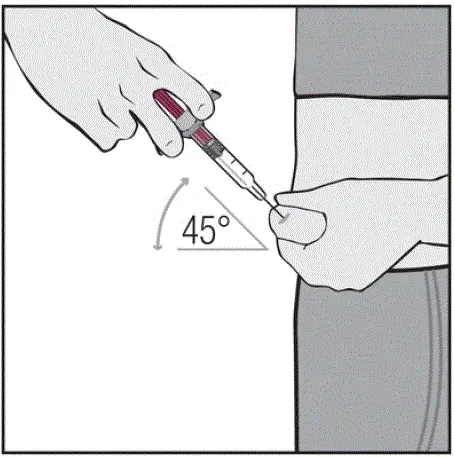
- Det betyder, at du er gået ind i et blodkar.
- Gør ikke inject Humira.
- Træk the needle out of the skin while keeping the syringe at the same angle.
- Trykke a cotton ball or gauze pad over the injection site og hold it for 10 sekunder. Se Figur j .
- Gør ikke Brug den samme sprøjte og nål igen. Kast nålen og sprøjte væk i din skarpe beholder.
- Gør ikke Gnid injektionsstedet. Du har muligvis let blødning. Dette er normalt.
- Gentag trin 1 til 12 med en ny præfyldt sprøjte.
Hvis der ikke vises noget blod i sprøjten:
- Skub langsomt stemplet helt ind, indtil al væsken injiceres, og sprøjten er tom.
- Træk the needle out of the skin while keeping the syringe at the same angle.
- Tryk på en bomuldskugle eller gasbind over injektionsstedet og hold den i 10 sekunder. Gør ikke Gnid injektionsstedet. Du har muligvis let blødning. Dette er normalt.







- Kast den brugte præfyldte sprøjte og nål i en skarp bortskaffelsesbeholder med det samme efter brug. Se Hvordan skal jeg smide (bortskaffe) brugte forudfyldte sprøjter og nåle?
- Hold en oversigt over datoer og placering af dine injektionssteder. For at hjælpe dig med at huske, hvornår du skal tage Humira, kan du markere din kalender på forhånd.
Hvordan skal jeg smide (bortskaffe) brugte forudfyldte sprøjter og nåle?
Figur k
 |
- Læg dine brugte nåle og sprøjter i en FDA-klaret skarp bortskaffelsescontainer med det samme efter brug. Se Figur k . Gør ikke throw away (dispose of) loose needles og syringes in your household tudslæt.
- Gør ikke try to touch the needle.
- Hvis du ikke har en FDA-Cleared Sharps bortskaffelsesbeholder, kan du bruge en husholdningsbeholder, der er:
- lavet af en kraftig plastik
- kan lukkes med et tæt passende punkteringsbestandigt låg uden at skarpe er i stand til at komme ud
- lodret og stabil under brug
- lækagebestandig og
- Korrekt mærket til at advare om farligt affald inde i beholderen.
- Når din Sharps -bortskaffelsescontainer er næsten fuld, skal du følge dine samfundsretningslinjer for den rigtige måde at bortskaffe din Sharps Disposal Container. Der kan være statslige eller lokale love om, hvordan du skal smide brugte nåle og sprøjter. For mere information om Safe Sharps bortskaffelse og for specifik information om Sharps bortskaffelse i den stat, du bor i, skal du gå til FDA's websted på: https://www.fda.gov/safesharpsdisposal.
- For sikkerheden og sundheden for dig og andre nåle og brugte sprøjter må aldrig genbruges.
- De brugte alkoholpuder bomuldskugler dosisbakker og emballage kan placeres i dit husholdningspass.
- Gør ikke dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Gør ikke recycle your used sharps disposal container.
- Hold altid skarpe beholder uden for børns rækkevidde.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet mellem 36 ° F (2 ° C (2 ° C (2 ° C). Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke Frys Humira. Gør ikke Brug Humira, hvis den er frosset, selvom det er optøet.
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -karton -dosisbakken eller præfyldt sprøjte. Gør ikke Brug Humira efter udløbsdatoen.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C 14 dage. Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Smid Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke blevet brugt indeni 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
- Gør ikke use a prefilled syringe if the liquid is cloudy discolorød or has flakes or particles in it.
- Gør ikke drop or crush Humira. The prefilled syringe is glass.
- Hold Humira -injektionsartikler og alle andre medicin uden for børns rækkevidde.
Brug til brug
Humira ®
(Hu-Mare-ah)
(adalimumab)
80 mg/0,8 ml 40 mg/0,4 ml 20 mg/0,2 ml og 10 mg/0,1 ml
Enkeltdosis forfyldt sprøjte
Før du injicerer: Din sundhedsudbyder skal vise dig, hvordan du bruger Humira, før du bruger den for første gang. Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Hvis du har brug for hjælp.
Figur a: Humira Single-Gørse Prefille
 |
Vigtige oplysninger, du har brug for at vide, før du injicerer Humira
Gør ikke Brug den forudfyldte sprøjte og ring til din sundhedsudbyder eller apotek, hvis:
- Væske er overskyet misfarvet eller har flager eller partikler i det
- Udløbsdato er gået
- Væske er frosset (selvom tøet) eller efterladt i direkte sollys
- Den forudfyldte sprøjte er blevet droppet eller knust
Opbevar nåledækslet indtil lige før din injektion.
Hvordan skal jeg opbevare Humira?
- Opbevar Humira i køleskabet mellem 36 ° F (2 ° C (2 ° C (2 ° C).
- Opbevar Humira i den originale karton, indtil den bruges til at beskytte den mod lys.
- Gør ikke freeze
- Kølet Humira kan bruges indtil udløbsdatoen, der er trykt på Humira -karton -dosisbakken eller præfyldt sprøjte.
- Hvis det er nødvendigt for eksempel, når du rejser, kan du også opbevare Humira ved stuetemperatur op til 25 ° C 14 dage.
- Smid Humira væk, hvis det er blevet holdt ved stuetemperatur og ikke brugt indeni 14 dage.
- Registrer den dato, hvor du først fjerner Humira fra køleskabet i de rum, der leveres på karton- og dosisbakken.
- Opbevar ikke Humira i ekstrem varme eller kulde.
Hold Humira -injektionsartikler og alle andre medicin uden for børn.
Læs instruktioner på alle sider, før du bruger Humira-enkeltdosis-præfyldt sprøjte
Tage Humira out of the refrigerator.
Forlade Humira at room temperature for 15 til 30 minutter inden injektion.
- Gør ikke Fjern nåledækslet, mens Humira tillader stuetemperatur
- Gør ikke Varm Humira på nogen anden måde. For eksempel Gør ikke Varm det i en mikrobølgeovn eller i varmt vand.
- Gør ikke Brug den forudfyldte sprøjte, hvis væske er frosset (selvom tøet)
 |
Check Udløbsdato på det forfyldte sprøjte etiket. Gør ikke Brug den forudfyldte sprøjte, hvis udløbsdatoen er gået.
Placere Følgende på en ren flad overflade:
- 1 enkeltdosis forfyldt sprøjte og alkoholpinde
- 1 bomuldskugle eller gasbindel (ikke inkluderet)
- Punkteringsbestandig Sharps bortskaffelsesbeholder (ikke inkluderet). Se trin 9 i slutningen af denne instruktioner til brug til instruktioner om, hvordan man smider væk (bortskaffer) din forudfyldte sprøjte
Vask og tør dine hænder.
 |
Vælge Et injektionssted:
- På fronten af dine lår eller
- Dit mave (mave) mindst 2 inches fra din navle (maveknap)
- Forskellig fra dit sidste injektionssted
Tørre Injektionsstedet i en cirkulær bevægelse med alkoholpinden.
- Gør ikke Injekt gennem tøj
- Gør ikke Injicer i huden, der er øm mærket rød hård arret har strækmærker eller områder med psoriasis plaques
 |
Holde den forudfyldte sprøjte i den ene hånd.
Træk forsigtigt Nåledækslet lige ud med den anden hånd.
- Kast nåledækslet væk
- Gør ikke Rør ved nålen med fingrene eller lad nålen røre ved noget
 |
Holde Den forudfyldte sprøjte med nålen vender op.
- Holde den forudfyldte sprøjte i øjenhøjde med den ene hånd, så du kan se luften i den forudfyldte sprøjte
- Brug af din anden hånd Skub langsomt Stemplet ind for at skubbe luften ud gennem nålen.
- Du kan se en dråbe væske i slutningen af nålen. Dette er normalt.
 |
Holde Kroppen af den forudfyldte sprøjte i den ene hånd mellem tommelfingeren og pegefingrene. Hold den forudfyldte sprøjte i din hånd som en blyant.
Gør ikke Træk tilbage på stemplet når som helst.
 |
Klem forsigtigt Området med renset hud på dit injektionssted med din anden hånd. Hold huden fast.
Indsæt Nålen ind i huden ved ca. en 45-graders vinkel ved hjælp af en hurtig dartlignende bevægelse.
- Efter nålen er i slip af huden.
Skub langsomt Stemplet hele vejen ind, indtil al væsken er injiceret, og den forudfyldte sprøjte er tom.
 |
Når injektionen afsluttes langsomt, skal du trække nålen ud af huden, mens du holder den forudfyldte sprøjte i samme vinkel.
Efter at have afsluttet injektionsstedet placerer en bomuldskugle eller gasbind på huden på injektionsstedet.
- Gør ikke gnide
- Let blødning på injektionsstedet er normalt
 |
Hvordan skal jeg bortskaffe den brugte Humira -præfyldte sprøjte?
- Læg dine brugte nålesprøjter og skarpe i en FDA-klaret skarp bortskaffelse af container med det samme efter brug. Gør ikke throw away (dispose of) loose needles og syringes in the household tudslæt.
- Hvis du ikke har en FDA-Cleared Sharps bortskaffelsesbeholder, kan du bruge en husholdningsbeholder, der er:
- lavet af en kraftig plastik
- kan lukkes med et tæt passende punkteringsbestandigt låg uden at skarpe er i stand til at komme ud
- lodret og stabil under brug
- lækagebestandig og
- Korrekt mærket til at advare om farligt affald inde i beholderen.
- Når din Sharps -bortskaffelsescontainer er næsten fuld, skal du følge dine samfundsretningslinjer for den rigtige måde at bortskaffe din Sharps Disposal Container. Der kan være statslige eller lokale love om, hvordan du skal smide brugte nåle og sprøjter. For mere information om Safe Sharps bortskaffelse og for specifik information om Sharps bortskaffelse i den stat, du bor i, skal du gå til FDA's websted på: https://www.fda.gov/safesharpsdisposal.
- Gør ikke dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Gør ikke recycle your used sharps disposal container.
 |
Nåledækslet alkoholpindekuglekugle eller dosisbakke og emballage kan placeres i dit husstands papirkurv.
Spørgsmål om brug af Humira-enkeltdosis-præfyldt sprøjte
Hvad hvis jeg ikke har modtaget en personuddannelse fra en sundhedsudbyder?
- Ring til din sundhedsudbyder eller 1-800-4Humira (1-800-448-6472) Eller besøg www.humira.com, hvis du har brug for hjælp
Hvad hvis jeg ikke har en FDA-Cleared Sharps bortskaffelsesbeholder eller korrekt husholdningsbeholder?
- Ring 1-800-4Humira (1-800-448-6472) For en gratis FDA-klaret skarp bortskaffelsesbeholder
Altid Hold den forudfyldte sprøjte og Sharps bortskaffelsesbeholder uden for børns rækkevidde.
Hold en oversigt over datoer og placeringer af dine injektioner. For at hjælpe med at huske, hvornår du skal tage Humira til at markere din kalender i forvejen.
 |
 |
Denne brugsanvisning er godkendt af U.S. Food and Drug Administration.